
Guide de préparations organiques à l’usage des étudiants/1
Gauthiers-Villars, (p. 1-82).
PREMIÈRE PARTIE.
1. Nitrobenzène C6H5.NO2.
Dans un ballon d’une contenance d’environ 300cm³ on mélange 150g d’acide sulfurique concentré et 100g d’acide nitrique ordinaire (de densité 1,41) ; on laisse refroidir à la température de la chambre et l’on ajoute par petites portions 50g de benzène, en agitant fréquemment et refroidissant par de l’eau.
Le ballon ne doit pas être bouché, car des gaz se dégagent pendant la réaction. Au cours de l’opération, le nitrobenzène se sépare du mélange acide sous la forme d’une couche huileuse.
Si une petite portion de cette dernière est versée dans de l’eau, elle doit tomber au fond, sinon il existe encore beaucoup de benzène non transformé. Une fois le benzène additionné, on continue d’agiter, tout en chauffant vers 60°, pendant une demi-heure à peu près.
La masse entière est alors versée dans environ 1 litre d’eau et bien remuée. L’huile épaisse qui se sépare est isolée par décantation au moyen d’un entonnoir à robinet. On lave l’huile encore une fois avec de l’eau, on la sépare le plus complètement possible de l’eau dans l’entonnoir à décantation et on la met dans un petit ballon de 100cm³ avec 5g à 10g de chlorure de calcium granulé.
Si l’on agite fréquemment le mélange, l’huile se trouve suffisamment séchée au bout de 12 heures. On la débarrasse du chlorure de calcium par filtration et on l’introduit dans un petit ballon à distiller, qui ne doit être rempli qu’à moitié environ par le liquide. Le fractionnement se fait dans l’appareil représenté par la figure 1. Il faut avoir soin de jeter dans le liquide deux ou trois fragments de porcelaine poreuse de la grosseur d’un pois.

Il passe à l’ordinaire premièrement de petites quantités de benzène et d’eau que l’on recueille séparément. Puis le thermomètre monte rapidement à environ 205° (non corr.) et le nitrobenzène distille dans l’intervalle de quelques degrés.
La distillation est interrompue dès que le contenu du ballon brunit fortement. On peut soumettre le nitrobenzène, pour le purifier complètement, à une seconde distillation. En opérant soigneusement, le rendement en nitrobenzène pur s’élève de 80 à 85 pour 100 du rendement théorique.
Si l’on procède comme il vient d’être dit, on trouve le point d’ébullition du nitrobenzène trop bas de quelques degrés, parce qu’une partie seulement de la colonne mercurique du thermomètre est chauffée par les vapeurs. L’appareil suffit néanmoins dans ce cas particulier où il s’agit de séparer le nitrobenzène du benzène non transformé ou du dinitrobenzène qui bout très haut.
Pour la détermination exacte du point d’ébullition on se sert de ballons à distiller à long col de la forme présentée par la figure 2 et de thermomètres courts (pour des corps à point d’ébullition élevé, on a recours aux thermomètres de Zincke). Pendant la distillation la colonne de mercure doit être plongée entièrement dans les vapeurs.

Le même appareil peut servir à la vérification des thermomètres ; on fait bouillir alors de l’eau, de la naphtaline, de la diphénylamine, etc. Il faut naturellement tenir compte de la hauteur barométrique.
2. Aniline C6H5.NH2.
Équation de formation :
Comme moyen de réduction on peut employer l’étain, le zinc, le fer et divers acides, ou du sulfure d’ammonium.
L’étain et l’acide chlorhydrique conviennent tout particulièrement pour des essais en petit, où le prix des matériaux n’entre pas directement en considération. On met 90g d’étain en grenailles et 50g de nitrobenzène dans un ballon d’une contenance approximative d’un litre et l’on ajoute par petites portions et en agitant fréquemment de l’acide chlorhydrique concentré. La masse s’échauffe passablement et il faut modérer la violence de la réaction en plaçant par moments le ballon dans de l’eau froide. La réaction est terminée lorsque l’odeur du nitrobenzène a complètement disparu.
Il arrive très souvent que pendant l’opération le sel double d’étain et d’aniline se sépare de la solution sous la forme d’une masse cristalline blanche. On ajoute alors à la fin de l’opération assez d’eau pour le dissoudre entièrement et l’on se débarrasse par décantation de l’étain non attaqué.
La liqueur acide est maintenant additionnée d’un excès de solution concentrée de soude caustique jusqu’à ce que les oxydes blancs d’étain, précipités au début, se redissolvent et que de l’étain métallique foncé se sépare. L’aniline se présente comme huile, que l’on peut extraire directement par l’éther ; cette méthode est d’une application générale pour tous les corps semblables. Il est préférable cependant de distiller en premier lieu la base avec les vapeurs d’eau.
On utilise pour cette opération le dispositif représenté par la figure 3. Le ballon a, contenant la solution d’aniline, est incliné de façon à éviter le rejaillissement du liquide dans le réfrigérant. Il n’est pas nécessaire de le chauffer. La vapeur, provenant d’une conduite ou produite dans un appareil quelconque, arrive par le tuyau b. L’aniline passe très facilement avec les vapeurs d’eau. On interrompt l’opération dès que le distillat s’écoule limpide. La couche huileuse d’aniline peut être siphonnée du distillat aqueux. Le rendement est cependant meilleur si l’on agite la totalité du distillat avec environ la moitié de son volume d’éther. Ceci peut se faire directement dans l’entonnoir à décantation. La solution éthérée est séparée, séchée avec du carbonate de potasse granulé, puis filtrée et évaporée dans un ballon au bain-marie. L’aniline reste sous la forme d’une huile limpide, jaune, que l’on fractionne de la même manière que le nitrobenzène. Le rendement est presque quantitatif. Point d’ébullition vers 181° (non corr.).

1. Essai avec le chlorure de chaux. On dissout une trace d’aniline dans de l’eau et l’on ajoute une solution filtrée de chlorure de chaux ; il se produit une coloration bleue violette intense.
L’essai sert également à reconnaître la présence de benzène qu’on transformera au préalable en aniline par les méthodes qui viennent d’être décrites.
2. Le sulfate est peu soluble. Quelques gouttes de la base sont additionnées d’acide sulfurique dilué, le sulfate se sépare et est recristallisé de l’eau chaude.
3. Formation de diazobenzène. On dissout quelques gouttes de la base dans de l’acide nitrique dilué, puis on ajoute quelques gouttes d’une solution de nitrite de soude : la liqueur reste limpide ; mais, si l’on chauffe, il se produit un fort dégagement de gaz et une huile brune se sépare, qui possède l’odeur caractéristique du nitrophénol.
4. Au mélange d’une goutte d’aniline et d’une goutte de chloroforme on ajoute une solution alcoolique de potasse et l’on chauffe. Aussitôt on perçoit l’odeur très désagréable de l’isonitrile phénylique. Aussi convient-il de faire l’essai sous une hotte.
3. Acétanilide C6H5.NH(C2H30).
Dans un ballon muni d’un réfrigérant ascendant on chauffe 20g d’aniline avec 30g d’acide acétique glacial pendant 6 à 10 heures, jusqu’à ce qu’une prise se cristallise par le refroidissement.
On verse en un mince filet le mélange encore chaud de la réaction dans environ 500cm³ d’eau chaude. La substance se dissout entièrement à l’ébullition, on ajoute à la solution une pointe de couteau de charbon animal et on laisse bouillir pendant 1 minute. On verse le liquide sur un filtre à plis humecté d’eau chaude et placé sur un entonnoir réchauffé. L’acétanilide, qui se sépare de la solution par le refroidissement, est essorée à la trompe, puis séchée dans l’exsiccateur à vide. Si elle n’est pas encore blanche, on répète la cristallisation dans l’eau bouillante, avec addition de charbon animal. La pureté du produit est contrôlée par le point de fusion. Dans ce but, on pulvérise une petite quantité de la substance séchée et on la met dans un tube capillaire. On se sert pour cette opération de l’appareil représenté par la figure 4. Le ballon est rempli aux quatre cinquièmes avec de l’acide sulfurique concentré. Le thermomètre, fixé à un bouchon placé légèrement sur l’orifice du ballon, plonge jusqu’au milieu du liquide. Le capillaire adhère au thermomètre mouillé par un peu d’acide sulfurique, de telle façon que la substance qui y est contenue se trouve à mi-hauteur du réservoir à mercure.

On emploiera toujours des thermomètres dits normaux[1].
Point de fusion 115° à 116°.
Réaction. — En chauffant l’acétanilide avec des alcalis, on perçoit l’odeur de l’aniline.
4. Sulfocarbanilide CS(NH.C6H5)2.
On introduit dans un ballon, muni d’un réfrigérant à reflux fonctionnant bien, 50g d’aniline, 50g d’alcool, 50g de sulfure de carbone et environ 0g,25 de soufre cristallisé. L’appareil est placé sous une hotte. On chauffe le tout au bain-marie en maintenant une douce ébullition pendant 5 ou 6 heures. Le mélange se prend peu à peu en une masse de lamelles cristallines. Pour finir, on distille le sulfure de carbone en se servant du réfrigérant descendant (prendre garde au danger d’inflammation du sulfure de carbone) ; le résidu est lavé à froid avec de l’acide chlorhydrique fortement dilué afin d’éliminer l’aniline non transformée. Les cristaux essorés sont dissous au bain-marie (réfrigérant ascendant) dans beaucoup d’alcool absolu. À la solution filtrée à chaud on ajoute juste assez d’eau chaude pour produire un léger trouble. Par refroidissement, la sulfocarbanilide cristallise, on l’essore et la sèche dans l’étuve. Le produit doit être complètement incolore et inodore ; on contrôle également la pureté par le point de fusion (152°). Rendement presque quantitatif.
5. Sulfocyanate de phényle C6H5.N = C:S
(Phénylsénévol).
Dans un ballon muni d’un réfrigérant ascendant et placé sous la hotte, on introduit 30g de sulfocarbanilide bien pulvérisée et trois fois autant d’acide chlorhydrique concentré. On laisse bouillir doucement pendant 45 minutes. La masse entière est ensuite versée dans l’eau. Le sénévol est chassé par un courant de vapeurs d’eau et le distillat épuisé par l’éther. On évapore le dissolvant, sèche le résidu sur du chlorure de calcium et l’on rectifie. Rendement : 10g.
Quelques gouttes du phénylsénévol additionnées d’une quantité égale d’aniline donnent immédiatement et avec une vive réaction, de la sulfocarbanilide.
Si l’on agite avec de l’ammoniaque concentrée, il se forme bientôt de la monophénylsulfocarbamide également solide.
6. β-phénylhydroxylamine C6H5.NH.OH.
On mélange 60g de nitrobenzène en suspension dans 1 litre et demi d’eau, avec 30g de chlorure d’ammonium. On ajoute au mélange, dans l’espace de 45 minutes, et en remuant fortement au moyen d’une turbine, peu à peu 80g de poudre de zinc. Il faut constamment observer la température et la maintenir par un refroidissement approprié entre 15° et 17°. Dans la règle, l’essai est achevé au bout d’une heure, ce que l’on constate à la disparition de l’odeur du nitrobenzène.
Le produit de la réaction est filtré aussi rapidement que possible à travers un filtre plissé. On additionne le filtrat de 500g de sel marin en poudre fine.
La β-phénylhydroxylamine se sépare alors immédiatement sous la forme d’une bouillie cristalline épaisse, composée de petites aiguilles incolores. Après un repos d’une demi-heure dans de la glace on essore et on lave les cristaux avec très peu d’eau froide.
Le produit ainsi obtenu est séché dans le vide, au-dessus de l’acide sulfurique. Rendement : 54g.
Pour purifier complètement le corps, on le recristallise dans du benzène chaud. Point de fusion : 81°. Réaction : réduction de la liqueur de Fehling.
7. Nitrosobenzène CH3.NO.
(Bamberqer, Ber. d. d. Chem. Ges., t. XXVII, p. 1555.)
On dissout, en remuant, 2g de β-phénylhydroxylamine pure et finement pulvérisée dans un mélange, refroidi à 0°, de 6g d’acide sulfurique concentré et 100cm³ d’eau. Puis on additionne d’un coup le mélange d’une solution préalablement préparée et également bien refroidie de 2g,4 de bichromate de potasse dans 150cm³ d’eau. Au bout de quelques instants la cristallisation du nitrosobenzène commence ; en laissant le produit de la réaction dans de la glace, la cristallisation est terminée après une demi-heure environ. On essore le nitrosobenzène, le lave avec de l’eau et le sèche dans le vide, au-dessus de l’acide sulfurique. Le rendement est presque quantitatif et le produit présente le point de fusion correct (68°).
Essai. — On chauffe un peu de nitrosobenzène dans un tube à essai avec un peu d’eau. À l’ébullition des gouttelettes huileuses vert d’émeraude et d’odeur excessivement piquante s’échappent avec les vapeurs d’eau et se prennent par le refroidissement en cristaux blanc de neige : le nitrosobenzène est en effet blanc à l’état solide, mais vert lorsqu’il est fondu ou en solution.
8. Benzoate d’éthyle C6H5.COOC2H5.
On ajoute 10g d’acide sulfurique concentré à une solution de 50g d’acide benzoïque dans 100g d’alcool absolu, et l’on maintient ce mélange à l’ébullition pendant 4 heures, le ballon étant pourvu d’un réfrigérant ascendant.
Pour finir, on distille à peu près la moitié de l’alcool au bain-marie, on dilue avec 300cm³ d’eau et l’on neutralise avec du carbonate de soude sec et pulvérisé, de façon à neutraliser tout l’acide sulfurique ainsi que l’acide benzoïque non transformé.
L’huile qui se sépare est extraite par de l’éther, la solution éthérée évaporée, et le résidu fractionné après avoir été séché sur du carbonate de potasse pur et calciné.
(Le carbonate de potasse utilisé doit être préparé en calcinant du bicarbonate pur.)
Point d’ébullition de l’éther : 212°. Rendement : 55g.
9. Acide m-bromobenzoïque C6H4Br.CO2H.
6g d’acide benzoïque, 8g de brome et à peu près 40g d’eau sont introduits dans un tube de verre résistant (tube à canon), dont l’une des extrémités est arrondie. On étire alors l’autre extrémité en un capillaire aussi épais que possible, puis on chauffe le tube dans un bain d’air (fourneau à canons) pendant à peu près 12 heures, de 140° à 150°. Comme il y a toujours de la pression dans de pareils tubes, on les ouvre de la manière suivante. On attend qu’ils se soient complètement refroidis, puis, en inclinant prudemment la gaine en fer forgé, on fait avancer le tube de verre jusqu’à ce que le capillaire sorte juste de l’orifice. On en chauffe l’extrême pointe jusqu’à fusion du verre. La pression, s’il y en a, souffle dans le capillaire une fine ouverture par où les gaz s’échappent. On opérera de manière à ce qu’en cas d’explosion les débris de verre ne puissent blesser personne. Aussitôt que les gaz ont entièrement disparu, le tube canon peut être retiré de la gaine métallique et coupé de la façon habituelle en éraflant avec le couteau à verre et faisant sauter.

Le brome doit avoir presque entièrement disparu, si l’opération a suivi son cours normal. On lave le produit de la réaction hors du tube, on le filtre et le triture dans une capsule avec de l’eau. Puis on le fait bouillir dans un ballon avec 500cm³ d’eau pendant environ 1 heure, afin d’éliminer complètement l’acide benzoïque non attaqué. Finalement la solution est traitée par un peu de charbon animal et filtrée à chaud. Par le refroidissement, l’acide bromobenzoïque se sépare en cristaux. On l’obtient absolument pur après une ou deux recristallisations dans l’eau chaude. Rendement : 7g. Point de fusion : 155°.
10. Chlorure de benzoyle C6H5.CO.Cl.
On place dans un ballon d’environ 500cm³, 50g d’acide benzoïque sec et 90g de pentachlorure de phosphore (ce dernier doit être pesé sous la hotte) et l’on secoue le mélange. Dans la règle, la réaction se produit d’elle-même ; si ce n’est pas le cas, on chauffe doucement.
La masse se liquéfie en dégageant beaucoup de vapeurs d’acide chlorhydrique.
La réaction est terminée aussitôt que tout l’acide benzoïque est entré en solution.
Le liquide représente alors un mélange de chlorure de benzoyle, d’oxychlorure de phosphore et d’un petit excès de pentachlorure de phosphore. On sépare ces divers produits par des distillations fractionnées répétées.
Le point d’ébullition du chlorure de benzoyle est situé à 199. Le rendement est presque quantitatif.
Le produit doit être conservé dans un flacon bien bouché afin d’être préservé de l’humidité de l’air.
1. Quelques gouttes de l’huile sont chauffées avec de l’eau jusqu’à dissolution ; par le refroidissement, il se sépare de l’acide benzoïque. La décomposition a lieu plus rapidement si l’on chauffe avec une solution de soude caustique ou de carbonate de soude ; il se forme alors du benzoate de soude facilement soluble.
2. Quelques gouttes de chlorure de benzoyle sont mélangées avec une quantité double d’alcool. On observe l’odeur du benzoate d’éthyle.
3. On mélange quelques gouttes du chlorure avec un léger excès d’aniline. La masse se prend, en dégageant beaucoup de chaleur, en un mélange de chlorhydrate d’aniline et de benzanilide. Cette dernière ne se dissout pas lorsqu’on lave avec de l’eau et peut être recristallisée dans l’alcool chaud.
11. Benzamide C6H5.CO.NH2.
On broie finement dans un mortier 15g de carbonate d’ammoniaque commercial (hotte), et l’on ajoute peu à peu, en remuant, 10g de chlorure de benzoyle. Si l’odeur de ce dernier ne disparaît pas complètement, ou prend encore quelques grammes de carbonate d’ammoniaque. La masse est lavée ensuite avec un peu d’eau froide pour enlever le chlorure d’ammonium et l’excès de carbonate. Le résidu essoré est recristallisé dans aussi peu d’eau chaude que possible. Rendement : environ 6g.
On peut employer, à la place de carbonate d’ammoniaque, une solution concentrée d’ammoniaque. Mais le rendement est alors moins bon. Point de fusion de la benzamide : 128°. En la chauffant avec des alcalis, elle dégage très rapidement de l’ammoniaque.
12. Nitrate de diazobenzène C6H5.N2.NO3.
À 20g d’aniline contenus dans un vase à précipiter on ajoute avec précaution, et en refroidissant bien, de l’acide nitrique du poids spécifique 1,41 préalablement bouilli et dilué avec la moitié de son volume d’eau. On s’arrête lorsque toute la masse se prend en une bouillie cristalline épaisse. Cette dernière est soigneusement essorée à la trompe et lavée avec un peu d’eau froide. On introduit dans un petit ballon environ 5g du sel humide et finement pulvérisé ; on ajoute juste assez d’eau pour baigner les cristaux. On refroidit dans de l’eau glacée, puis on fait passer dans le mélange de l’acide nitreux gazeux. Celui-ci est obtenu dans le ballon a (fig. 8) à partir d’acide arsénieux en morceaux et d’acide nitrique ordinaire. En passant par le flacon vide b il abandonne l’acide nitrique, etc.

On arrive à régler le courant gazeux à volonté en chauffant doucement et en refroidissant par de l’eau froide alternativement le ballon a.
La température dans le ballon c, qu’on remue fréquemment, ne doit pas dépasser 10°. On poursuit l’introduction des vapeurs rouges, jusqu’à ce que tout le nitrate d’aniline ait disparu. Généralement, celui-ci se dissout complètement en donnant une solution limpide. Mais, si la quantité d’eau n’est pas suffisante, une partie du nitrate de diazobenzène peut déjà se séparer pendant l’opération. La forme cristalline de ce dernier permet cependant de le distinguer très facilement du sel d’aniline. La réaction une fois terminée, on verse le contenu de c dans trois fois son volume d’alcool absolu et l’on ajoute de l’éther tant qu’il se précipite des aiguilles blanches.
Si, dès le début, on a additionné le nitrate d’aniline d’un excès d’eau, il ne se sépare point de cristaux, mais bien une solution aqueuse épaisse de nitrate de diazobenzène. Il est alors nécessaire, après avoir décanté la partie éthéro-alcoolique, de dissoudre de nouveau la solution aqueuse dans de l’alcool absolu et de reprécipiter par de l’éther. Le nitrate de diazobenzène précipité est un explosif dangereux à l’état sec ; on l’essore à la trompe, le lave avec de l’éther, et le dissout encore humide dans de l’eau glacée. On sépare la solution aqueuse de la couche éthérée en siphonnant et l’on se sert de la première pour les essais suivants :
1. Si l’on chauffe le liquide, il se produit un dégagement tumultueux d’azote, et une huile brune, sentant fortement le nitrophénol, se sépare. Le nitrophénol résulte de l’action de l’acide nitrique devenu libre sur le phénol qui se forme d’abord. C’est pourquoi l’on n’emploie pas les nitrates, mais bien les sulfates, pour préparer les phénols à partir des amines aromatiques.
2. En ajoutant à la solution de nitrate de diazobenzène un sel d’aniline et de l’acétate de soude en excès, il se précipite du diazoamidobenzène sous la forme de petits cristaux jaunes.
3. Si l’on additionne la solution de diazobenzène d’une solution acétique de diméthylaniline, il se produit, au bout de peu de temps, une magnifique coloration rouge (formation d’une matière colorante azoïque).
4. Quand on verse dans la solution de diazobenzène une solution de brome dans de l’acide bromhydrique, il se sépare une huile brun rouge. Celle-ci se prend bientôt en lamelles cristallines, lorsque, après décantation de la couche aqueuse, on la lave avec un peu d’éther. Ces cristaux sont du perbromure de diazobenzène C6H5N2Br3. Si l’on en a une quantité suffisante, on peut l’employer pour la préparation du diazobenzènimide. Dans ce but, on recouvre les cristaux d’ammoniaque concentrée. Il se produit une vive réaction et les cristaux se transforment en une huile foncée, d’odeur caractéristique, stupéfiante, formée en majeure partie de diazobenzènimide.
13. Diazoamidobenzène C6H5.N = N — NH.C6H5.
On dissout 10g d’aniline dans 100g d’eau et la quantité d’acide chlorhydrique concentré calculée pour 2mol,5 (la concentration de l’acide doit être déterminée au moyen de l’aréomètre). Ensuite on refroidit soigneusement dans l’eau glacée, et l’on transforme l’aniline en chlorure de diazobenzène en ajoutant la quantité de nitrite de soude calculée exactement pour 1molet dissoute dans peu d’eau[2].
On doit soigneusement éviter un excès de nitrite (?), tandis qu’une quantité insuffisante n’affecte pas la pureté du diazoamidobenzène, mais seulement le rendement (?). Entre temps, on dissout 10g (ou mieux 11g) d’aniline dans 50g d’eau et la quantité d’acide chlorhydrique strictement nécessaire (au plus 1mol d’acide chlorhydrique pour 1mol d’aniline) ; on refroidit bien la liqueur et on la mélange à la solution du chlorure de diazobenzène, puis on ajoute au tout environ 40g d’acétate de soude, dissous dans très peu d’eau.
Le diazoamidobenzène se sépare sous la forme d’un précipité cristallin d’un beau jaune. Si le produit est jaune foncé ou brun, c’est qu’il y a un excès de diazobenzène, ou qu’on n’a pas suffisamment refroidi.
Le dépôt du diazoamidobenzène prend un certain temps ; lorsque, après environ 30 minutes, une prise d’essai ne donne plus de précipité cristallin par addition d’acétate de soude, on essore le précipité à la trompe, lave soigneusement avec de l’eau froide, et l’on enlève l’eau aussi complètement que possible par expression. La masse est alors immédiatement dissoute dans de la ligroïne chaude, bouillant entre 70 et 100°, la solution étant filtrée chaude pour enlever l’eau en suspension. Par refroidissement, le diazoamidobenzène se sépare sous la forme de cristaux jaune foncé, bien formés. Ils sont bruns, si la préparation a été défectueuse. Rendement : 15g.
14. Amidoazobenzène C6H5.N = N.C6H4.NH2
On dissout 10g de diazoamidobenzène dans 20g d’aniline et l’on ajoute 5g de chlorhydrate d’aniline solide. On chauffe ce mélange de 40° à 50°, jusqu’à ce qu’un échantillon porté à l’ébullition avec de l’alcool et de l’acide chlorhydrique ne donne plus de dégagement gazeux (30 minutes à 1 heure). On verse le liquide dans un excès d’acide acétique très dilué. La base se précipite, et, après solidification, on l’essore et la lave avec de l’eau. On fait alors bouillir le précipité avec environ 2l d’eau et l’on ajoute prudemment de l’acide chlorhydrique jusqu’à ce qu’une prise d’essai de la solution bleu rouge dépose par refroidissement des cristaux d’un bleu pur. À ce moment, on filtre et laisse refroidir ; le chlorhydrate de l’amidoazobenzène se sépare sous la forme de petites aiguilles d’un bleu d’acier. On peut précipiter le reste en ajoutant du sel marin.
Pour préparer la base libre, on chauffe le chlorhydrate avec deux fois son poids d’alcool et l’on ajoute de l’ammoniaque concentrée jusqu’à ce que tout soit dissous et que la couleur ait passé au brun clair.
L’addition prudente d’eau précipite la base sous forme de beaux petits cristaux jaunes, que l’on peut recristalliser dans de l’alcool dilué. Rendement environ 8g.
| 15. Acide sulfanilique C6H4 | NH2 (1) |
| SO3H (4) |


On verse peu à peu et en remuant bien 50g d’aniline dans 150g d’acide sulfurique fumant (contenant 8 à 10 pour 100 d’anhydride)[3], puis on chauffe le mélange au bain d’huile à 170° pendant 3 à 4 heures. La réaction est terminée lorsqu’une prise d’essai additionnée d’eau et d’un léger excès de soude ne donne plus d’aniline. Toute la masse est alors versée dans à peu près un tiers de litre d’eau glacée ; l’acide sulfanilique se précipite à l’état cristallin. Suivant le degré de pureté de l’aniline et la façon de chauffer, l’acide est plus ou moins coloré. Pour le purifier, on le dissout dans un peu plus que la quantité calculée[4] de soude caustique très diluée (le sulfanilate de soude est difficilement soluble dans la soude concentrée), on fait bouillir avec du charbon animal et l’on précipite par un acide dilué après filtration.
Si le produit est encore coloré, on répète le traitement avec le charbon animal. Rendement de 55g à 60g.
Le sulfanilate de soude est le plus beau des sels de l’acide sulfanilique. Il précipite par refroidissement d’une solution concentrée de l’acide dans de la soude étendue, chaude, en cristaux incolores, bien formés.
| 16. Acide diazobenzènesulfonique C6H4 | N = N |
| SO3 |
On dissout à chaud 20g d’acide sulfanilique séché au bain-marie et finement pulvérisé dans la quantité calculée de soude caustique et l’on dilue la solution de façon qu’il ne se produise pas de cristallisation en refroidissant à 50°. On ajoute, à cette température, un peu plus que la quantité calculée de nitrite de soude et l’on verse le mélange en agitant bien dans un excès d’acide sulfurique dilué et refroidi. Le dérivé diazoïque se précipite bientôt en une masse cristalline, incolore. On favorise la cristallisation en refroidissant et l’on filtre après quelque repos.
Le produit est relativement stable ; il peut même être recristallisé dans l’eau à 60°. Il se dissout aussi sans décomposition dans les alcalis et en est reprécipité par les acides. Ce corps peut être conservé à l’état sec, mais ne doit pas être séché à 100°. Du reste, le produit sec ne doit être manié qu’avec prudence, car il fait parfois explosion par simple trituration. Bon rendement.
17. Hélianthine HSO3.C6H4.N = N.C6H4.N(CH3)2
On dissout l’acide diazobenzène sulfonique obtenu par le procédé indiqué ci-dessus dans la plus petite quantité possible d’une solution de soude caustique diluée et en refroidissant avec de la glace. Puis on verse le liquide dans de l’acide acétique contenant en solution la quantité calculée de diméthylaniline. Le sel de soude de la matière colorante se sépare alors en petites paillettes brillantes, de couleur jaune orange.
Celles-ci sont essorées et recristallisées dans l’eau chaude. Si l’on additionne leur solution d’acide acétique dilué, la matière colorante libre se précipite sous la forme d’une poudre cristalline orangée, tandis qu’un excès d’acide chlorhydrique donne le chlorhydrate formant de beaux cristaux violets. Il n’est pas nécessaire, pour préparer la matière colorante, d’isoler la combinaison diazoïque. On peut partir directement de l’acide sulfanilique et effectuer, en une seule opération, la diazotation et la formation de la matière colorante.
On opère de la manière suivante :
On dissout 1mol (10g) d’acide sulfanilique dans exactement 1mol de soude caustique en solution diluée, on ajoute 1mol NaNO2 et à froid 1mol d’acide chlorhydrique. Cette solution est additionnée directement de 1mol de diméthylaniline en solution dans un peu d’acide chlorhydrique. On ajoute alors de nouveau de la soude caustique. Au bout de peu de temps, le sel de soude de la matière colorante se précipite. On achève la précipitation en dissolvant du sel marin dans la liqueur.
18. Phénylhydrazine C6H5.NH−NH2.
1. On dissout 50g d’aniline dans 2mol,5 d’acide chlorhydrique concentré et 300g d’eau, on refroidit bien et l’on diazote au moven de la quantité calculée de nitrite de soude. On verse le tout dans une solution froide et à peu près saturée de 2mol,5 Na2SO3.
On emploie pour cela la solution de bisulfite de soude du commerce qui contient environ 40 pour 100 de NaHSO3 et qu’on neutralise par une solution de soude caustique.
Une prise d’essai du liquide doit rester limpide à l’ébullition sinon la quantité de sulfite serait insuffisante. On chauffe alors au bain d’air, (sous la hotte) la solution contenue dans un grand ballon à fond rond ; on ajoute de la poudre de zinc et un peu d’acide acétique jusqu’à ce qu’elle soit devenue incolore. On sépare le liquide encore chaud du zinc par filtration. Dans le filtrat se trouve le phénylhydrazine sulfonate de soude ; on l’additionne immédiatement et à chaud d’un tiers de son volume d’acide chlorhydrique fumant (attention !). Le produit se prend en une masse cristalline de chlorhydrate de phénylhydrazine. Après refroidissement de la solution, on essore soigneusement le sel et on le sépare autant que possible des eaux mères par expression.
Ensuite on recouvre le sel d’un excès de solution de soude et l’on agite ; la base qui se sépare est extraite par de l’éther. On dessèche la solution éthérée pendant 12 heures au contact de carbonate de potasse ; on filtre, on évapore et l’on distille le résidu dans le vide en se servant de l’appareil représenté dans la figure 9 : a et b sont des ballons à distiller. Le premier se trouve dans un bain d’huile et doit être rempli au plus jusqu’au tiers. Pour faciliter l’ébullition, on laisse passer pendant l’opération et au moyen d’un capillaire, un faible courant d’air réglé par la pince c. Le second ballon est refroidi par un jet d’eau. Il suffit, pour que la phénylhydrazine distille, sous une pression de 12mm, de chauffer le bain d’huile entre 120° et 140°.

2. On dissout 10g d’aniline dans 100cm³d’acide chlorhydrique concentré ; on diazote en ajoutant la quantité calculée de nitrite de soude. La liqueur est ensuite versée lentement et en remuant bien dans une solution refroidie et fortement acide de chlorure stanneux (préparée en dissolvant 60g de chlorure stanneux commercial dans de l’acide chlorhydrique). Le chlorhydrate de phénylhydrazine, qui se sépare aussitôt, est ensuite traité comme il est indiqué ci-dessus.
1. La phénylhydrazine réduit la liqueur de Fehling déjà à la température ordinaire.
2. Elle le forme avec l’acide chlorhydrique un sel difficilement soluble.
19. Nitrile benzoïque C6H5.CN.
Les opérations qui suivent, jusqu’à la distillation finale avec les vapeurs d’eau, doivent être exécutées sous une hotte à bon tirage.
On prépare en premier lieu une solution de cyanure cuivreux. Pour cela, on dissout dans un ballon de 2l de capacité 5g de sulfate de cuivre dans 200cm³ d’eau. À cette solution chaude on ajoute peu à peu une solution de 50g de cyanure de potassium dans 100cm³ d’eau. Il se produit tout d’abord un vif dégagement gazeux (cyanogène, toxique) et un précipité qui se redissout plus tard. On ajoute peu à peu à cette liqueur, chauffée à 70° environ, une solution de chlorure de diazobenzène que l’on a préparée préalablement en diazotant une solution de 19cm³ (= 18g,6) d’aniline dans 160cm³ d’eau et 40cm³ d’acide chlorhydrique concentré, avec 15g de nitrite de soude dans 40cm³ d’eau. Ceci fait, on chauffe pendant 15 minutes au bain-marie ; le nitrile se sépare sous la forme d’une huile foncée surnageante. On chasse celle-ci par un courant de vapeurs d’eau et l’on épuise le distillat par l’éther. La solution éthérée est, à plusieurs reprises, fortement secouée, d’abord avec de la soude caustique diluée, puis avec de l’acide sulfurique dilué. Elle est enfin séchée par du carbonate de potassium calciné. La solution filtrée laisse, après évaporation de l’éther, une huile brune que l’on purifie par fractionnement. Le rendement est environ de 13g.
20. Monoéthylaniline C6H5.NH.C2H5.
On chauffe au bain-marie, dans un ballon muni d’un réfrigérant ascendant, 50g d’aniline et 65g de bromure d’éthyle (un peu plus que la quantité calculée). On maintient à une douce ébullition, pendant 1 à 2 heures, jusqu’à ce que la masse se solidifie presque entièrement. On dissout ensuite dans l’eau, on chasse par ébullition la petite quantité de bromure d’éthyle non transformé. Ensuite on sursature la solution avec de la soude caustique et l’on extrait par l’éther les bases qui se sont séparées. L’huile qui reste après évaporation de l’éther représente un mélange d’aniline non transformée, de mono- et de diéthylaniline. On la dissout dans un excès d’acide chlorhydrique dilué (100g d’acide chlorhydrique fumant et 500g d’eau), on refroidit avec de la glace et l’on ajoute environ 37g de nitrite de soude. Il se forme du chlorure de diazobenzène, du chlorhydrate de nitrosodiéthylaniline et de l’éthylphénylnitrosamine. Cette dernière se sépare sous la forme d’une huile foncée que l’on extrait immédiatement par l’éther.
Pour régénérer l’éthylaniline à partir de la nitrosamine on réduit celle-ci, après évaporation de l’éther, au moyen d’étain et d’acide chlorhydrique, comme pour le nitrobenzène. La base est ensuite isolée de la solution chlorhydrique de la même manière que pour l’aniline et distillée, après avoir été desséchée sur de la potasse. Point d’ébullition : 204°. Rendement de 20g à 25g.
| 21. Nitrodiméthylaniline C6H4 | NO |
| N(CH3)2 |
On dissout 20g de diméthylaniline dans 100g d’acide chlorhydrique à 20 pour 100. Dans la solution bien refroidie on laisse tomber goutte à goutte et en agitant la quantité calculée de nitrite de soude dissoute dans peu d’eau. Le chlorhydrate du dérivé nitrosé se sépare déjà pendant l’opération sous forme d’aiguilles jaunes. Pour achever la cristallisation, on laisse reposer environ une heure, puis on essore à la trompe et on lave avec de l’acide chlorhydrique dilué. Le sel peut être facilement purifié par recristallisation dans l’eau chaude. Si l’on veut avoir la base libre, on suspend le sel dans l’eau, puis l’on ajoute de la soude caustique à froid et la base séparée est extraite par l’éther. La combinaison se précipite par évaporation de l’éther sous la forme de magnifiques paillettes jaune vert.
Rendement presque quantitatif.
1. La solution chlorhydrique de la base est rapidement décolorée par de l’étain ou du zinc (formation de paraamidodiméthylaniline).
2. Chauffée à l’ébullition avec une solution de soude caustique, la combinaison nitrosée dégage l’odeur de diméthylamine ; en même temps, la solution alcaline devient rouge foncé grâce à la formation de nitrosophénol.
22. Hydrazobenzène C6H5.NH — NH.C6H5.
On réduit le nitrobenzène par de la soude caustique et de la poudre de zinc. Pour accélérer la dissolution du premier on ajoute un peu d’alcool.
Comme la réaction est vive, il ne faut ajouter la poudre de zinc que peu à peu. Une forte agitation est nécessaire pour que la réaction marche bien. L’alcool qui distille pendant l’opération doit être condensé au moyen d’un réfrigérant.
On réalise aisément ces conditions en utilisant l’appareil suivant.

Le ballon a, à fond rond, mesure environ 1l,5 ; le diamètre de son col est d’à peu près 4cm et supporte l’agitateur grâce à un bouchon de caoutchouc. La forme d’agitateur indiquée par Schultze est très efficace et se prête particulièrement bien à l’introduction dans le ballon. Au repos, les ailes pendent verticalement et se tiennent horizontales lorsqu’elles sont en mouvement. L’agitateur b est relié à une petite turbine hydraulique portative fonctionnant bien ou à un petit électromoteur. Il doit faire 400 à 500 tours à la minute pendant le processus de la réduction, de sorte que le liquide se trouve fortement remué. La tige de l’agitateur passe par un tube de verre c, fixé au grand ballon par un bouchon en caoutchouc. Pour qu’aucune vapeur ne s’échappe du ballon par le tube, celui-ci porte une manchette d, contenant un peu d’eau. Une fermeture très efficace pour les vapeurs montantes résulte du fait que la tige de l’agitateur est reliée par un bouchon de caoutchouc à un petit cylindre de verre e, qui plonge et se meut dans l’eau. Deux tubes sont soudés au col du grand ballon et forment un angle d’à peu près 45° avec lui. L’un des deux, f, a environ 2cm de diamètre et 2cm à 3cm de longueur. Il est fermé par un bouchon et sert à l’introduction de la poudre de zinc. L’autre tube, g, long de 5cm à peu près, est surmonté d’un réfrigérant. On introduit dans le grand ballon 50g ou 42cm³ de nitrobenzène, 180cm³ de soude caustique (30 pour 100), 20cm³ d’eau et 50cm³ d’alcool. Puis, tandis que le mélange est vivement remué par l’agitateur, on jette par le tube f (le mieux au moyen d’un petit tube à produit) à peu près 10g de poudre de zinc. La liqueur s’échauffe bientôt. On poursuit l’addition de la poudre de zinc, mais par plus petites quantités (3g à 4g), jusqu’à ce que le liquide entre en ébullition. Comme la réaction peut devenir assez vive, et que la masse peut écumer fortement et remplir entièrement le ballon, on attendra toujours que l’écume ait disparu avant d’ajouter une nouvelle portion de poudre de zinc. Le danger du débordement de l’écume diminue de plus en plus vers la fin de la réaction. On peut facilement conduire l’opération de telle sorte qu’elle soit terminée au bout de 45 minutes à peu près. On reconnaît la fin au changement de coloration du liquide qui, franchement rouge au commencement, devient faiblement jaunâtre à la fin. Pour s’en rendre compte, on filtre une petite prise d’essai, extraite au moyen d’une pipette.
La quantité de poudre de zinc nécessaire varie avec sa composition. En général, 100g à 125g suffisent. Après avoir introduit toute la poudre de zinc, on continue d’agiter pendant environ 15 minutes, on ajoute alors à peu près 1l d’eau froide et l’on filtre à la trompe le mélange de poudre de zinc et d’hydrazobenzène précipité. Après avoir éliminé l’alcali en lavant avec de l’eau et après avoir essoré soigneusement, on chauffe la masse au bain-marie avec environ 0l,75 d’alcool pour dissoudre l’hydrazobenzène. La solution est filtrée à chaud, puis entourée d’un mélange réfrigérant. La plus grande partie de l’hydrazobenzène se dépose en une masse cristalline, composée de paillettes brillantes presque incolores. Les eaux-mères servent à épuiser encore une fois le résidu de zinc et sont additionnées à chaud de beaucoup d’eau chaude, ce qui provoque la cristallisation du restant de l’hydrazobenzène. Ces cristaux sont aussi bien formés, mais un peu colorés en orange. Le rendement total comporte environ 33g, soit les 88 pour 100 de la théorie. On peut obtenir le produit tout à fait incolore et fondant entre 125° et 126° par recristallisation dans l’alcool chaud. Il est préférable de le conserver dans un tube scellé, car il se colore lentement à l’air, en s’oxydant.
La manière de se comporter de l’hydrazobenzène vis-à-vis de la solution de Fehling est caractéristique. De même que la phénylhydrazine, il la réduit fortement à chaud.
Si l’on veut éviter le dispositif d’agitation décrit ci-dessus, très efficace, mais un peu compliqué, on utilisera un ballon à fond rond de 1l,5 ne portant que le tube latéral f pour l’introduction de la poudre de zinc ; le col du ballon est fermé par un bouchon par lequel passe un tube de verre assez large. Ce dernier est relié par un court tuyau de caoutchouc à un réfrigérant, de telle façon que l’on puisse secouer fortement le ballon avec la main pendant la réduction. L’introduction de la poudre de zinc s’effectue par le tube latéral de la manière indiquée. Mais, comme l’agitation à la main n’est pas aussi régulière, la marche de la réaction s’en ressent et le rendement est, en conséquence, un peu plus faible.
La transposition de l’hydrazobenzène en benzidine NH2.C6H4.C6H4.NH2 s’effectue le mieux au moyen d’acide très dilué et à basse température. On secoue 5g d’hydrazobenzène finement pulvérisé avec 125cm³ d’acide chlorhydrique à 3 pour 100. On maintient la température entre 20° et 30°. La dissolution est presque complète au bout de 15 à 30 minutes, si le produit de départ était pur. Pour finir, on chauffe quelque temps entre 45° et 50° et l’on ajoute encore un peu d’eau afin de dissoudre le sel de benzidine qui peut avoir cristallisé. On filtre à chaud. L’addition d’acide sulfurique à la solution, à peine colorée, précipite le sulfate. Si l’on emploie l’acide chlorhydrique concentré, on a le chlorhydrate. Les deux sels constituent une masse cristalline, incolore. Pour obtenir la base libre, on sursature la solution froide des sels avec une solution de soude caustique, on filtre à la trompe et on lave avec de l’eau froide. Rendement environ 85 pour 100. Pour purifier complètement la benzidine, on la recristallise dans beaucoup d’eau chaude, ou dans très peu d’alcool chaud, Point de fusion vers 125°. Contrairement à l’hydrazobenzène, la base ne réduit pas la solution de Fehling.
23. Iodure d’éthyle C2H5.I.
On recouvre 10g de phosphore amorphe, placé dans un ballon, avec 50g d’alcool absolu ; puis on ajoute dans l’intervalle de 1 heure à 1 heure et demie, en agitant fréquemment, 100g d’iode pulvérisé. On laisse séjourner plusieurs heures le mélange à la température ordinaire, tout en le secouant de temps en temps. Puis on chauffe avec réfrigérant ascendant pendant une heure au bain-marie ; enfin on distille, toujours au bain-marie, la plus grande partie du liquide.
Le résidu se compose de phosphore amorphe et d’une solution concentrée d’acides phosphoreux et phosphorique, qu’on ne peut pas utiliser.
Le distillat, coloré en brun par de l’iode libre, est un mélange d’alcool et d’iodure d’éthyle. On l’additionne de plusieurs fois son volume d’eau et de suffisamment de solution de soude caustique pour qu’en agitant fortement, l’iodure d’éthyle séparé se décolore complètement.
On décante ensuite l’huile au moyen d’un entonnoir à robinet ; on la sèche avec du chlorure de calcium granulé et l’on distille en présence de celui-ci, au bain-marie[5].
Si l’on a éloigné complètement l’alcool par des lavages le produit est chimiquement pur. L’iodure d’éthyle conservé dans des flacons en verre se colore peu à peu en violet et en brun, par des traces d’iode qui se séparent. On peut éviter ceci en ajoutant au liquide une petite quantité d’argent métallique finement divisé.
Rendement : 100g.
24. Aldéhyde CH3.CHO et Aldéhyde-ammoniaque CH3.CHO.NH3.
On introduit 200g de bichromate de potasse, en morceaux de la grosseur d’une lentille, dans un ballon d’au moins 2l de capacité, muni d’un réfrigérant, qui est relié à un récipient placé dans un mélange réfrigérant. On ajoute 600g d’eau, puis on laisse couler lentement et en remuant fréquemment un mélange de 200g d’alcool et 270g d’acide sulfurique concentré, placé dans un entonnoir à robinet (dit à brome). La masse s’échauffe d’elle-même, se colore en vert, et l’aldéhyde distille en quantité assez abondante, à côté d’alcool et d’eau.
Vers la fin on chauffe pour faire passer complètement le reste de l’aldéhyde.
Le distillat est redistillé d’un ballon a (fig. 11) se trouvant sur un bain-marie. Les vapeurs passent dans le réfrigérant ascendant d, rempli d’eau à 25° prise dans le vase e au moyen du tuyau de caoutchouc f. L’écoulement de l’eau se fait par le tuyau g et se règle par la pince à vis h.
Les vapeurs d’alcool et d’eau se condensent dans le réfrigérant, tandis que les vapeurs d’aldéhyde passant par le tube i et l’entonnoir b arrivent dans le récipient c. Celui-ci contient de l’éther sec, refroidi, qui absorbe facilement l’aldéhyde. Si l’on fait ensuite passer du gaz ammoniac sec[6] dans l’éther convenablement refroidi, l’aldéhyde-ammoniaque se sépare immédiatement en cristaux. On essore ces derniers à la trompe et on les sèche sur du papier à filtrer, après les avoir lavés avec de l’éther. Pour obtenir de l’aldéhyde pure, on distille les cristaux en présence d’acide sulfurique dilué. L’aldéhyde est séchée sur du chlorure de calcium et redistillée. Point d’ébullition : 21°.

Il sera avantageux d’utiliser immédiatement l’aldéhyde-ammoniaque fraîchement obtenue pour la préparation 57.
1. Réduction d’une solution ammoniacale d’argent : miroir d’argent. La réaction peut ne pas réussir en présence d’un excès d’ammoniaque.
2. Coloration d’une solution aqueuse d’aldéhyde lorsqu’on la chauffe avec des alcalis.
3. Transposition en paraldéhyde par une goutte d’acide sulfurique concentré.
25. Bromure d’éthylène C2H4Br2.
On mélange, dans le ballon a d’une capacité de 2l environ (fig. 12), 50g d’alcool absolu avec 300g d’acide sulfurique concentré. (Dans le dessin, le ballon est représenté trop petit.) On chauffe prudemment le mélange jusqu’à qu’il se produise un vif dégagement d’éthylène.

On y laisse alors arriver, par un entonnoir à robinet, dont l’extrémité est effilée, un mélange formé d’une partie d’alcool et de deux parties d’acide sulfurique concentré. L’écoulement doit être suffisamment rapide pour que le dégagement gazeux soit constant, sans qu’il y ait une mousse trop abondante. Les gaz qui s’échappent traversent les deux flacons laveurs b et c, contenant le premier de l’eau, le second une solution diluée de soude caustique. Ils sont ensuite reçus dans le flacon à absorption d, qui contient 100g de brome et qui est refroidi par de la glace. Le dégagement d’éthylène est poursuivi jusqu’à ce que le brome soit entièrement décoloré, ce qui nécessite parfois une nouvelle charge de l’appareil producteur d’éthylène. Ensuite le contenu du flacon d est lavé avec une solution étendue de soude caustique ; l’huile est décantée, séchée sur du chlorure de calcium, filtrée de ce dernier, puis fractionnée. Point d’ébullition : 131°,5. Rendement : 100g.
26. Glycol C2H4(OH)2.
On chauffe au réfrigérant ascendant, pendant environ 10 heures, 94g de bromure d’éthylène, 69g de carbonate de potassium pur (préparé à partir du bicarbonate) et 500g d’eau. Au bout de ce temps, tout le bromure aura disparu. Il est bon de placer des baguettes de bois dans le ballon pour faciliter l’ébullition et le mélange de l’huile avec l’eau.
Le bromure d’éthylène disparu, on concentre dans le vide la solution aqueuse, chauffée dans un bain d’eau à 50°. On utilisera l’appareil décrit pour la préparation de la phénylhydrazine. La masse cristalline restante paraît presque sèche ; on épuise à deux reprises par de l’alcool absolu, on filtre et l’on concentre le filtrat alcoolique de nouveau dans le vide. Le résidu est fractionné à la pression ordinaire, le mieux dans un bain d’huile.
Point d’ébullition du glycol : 197°. Rendement : 9g à 10g.
27. Méthylamine CH3.NH2.
On introduit dans un ballon d’un demi-litre 25g d’acétamide débarrassée de l’huile par expression sur une plaque poreuse ou entre deux feuilles de papier à filtrer au moyen d’une presse à main. On les recouvre de 23cm³ de brome et l’on ajoute peu à peu et en refroidissant bien avec de l’eau glacée, suffisamment d’une solution de 4g d’hydrate de potassium dans 350cm³ d’eau, pour que la liqueur devienne jaune clair. On verse celle-ci dans une solution de 80g de potasse caustique dans 150cm³ d’eau, contenue dans un ballon de 1l et préalablement chauffée à environ 75°. Puis on chauffe prudemment au bain-marie en contrôlant avec un thermomètre, jusqu’à ce que la température atteigne environ 70°. Le liquide s’échauffe maintenant de lui-même et il faut prendre garde de refroidir à temps, de façon que la température ne dépasse pas 75°. Aussitôt que la solution, maintenue de 70° à 75°, est devenue limpide et incolore, et qu’on n’observe plus d’élévation de température en l’absence de toute source de chaleur extérieure, on laisse digérer quelque temps à la même température. On distille ensuite l’amine formée en utilisant un réfrigérant descendant ; l’opération se poursuit jusqu’à ce que le distillat ne soit plus alcalin. Il est bon de munir l’extrémité du réfrigérant d’une allonge large, courbée vers le bas et qui plonge environ à 1cm de profondeur dans de l’acide chlorhydrique dilué. On peut aussi se servir du dispositif indiqué à la préparation n° 24 (aldéhyde-ammoniaque) pour éviter que l’acide ne remonte. La distillation terminée, on évapore la solution acide, on sèche le résidu pulvérisé à 110° environ et on l’épuise avec à peu près 15 fois son poids d’alcool absolu chaud. Le chlorure d’ammonium ne se dissout pas, tandis que le chlorhydrate de méthylamine cristallise rapidement dans le filtrat en paillettes minces, brillantes. On fera bien d’essorer la solution à la trompe, le flacon à vide étant préalablement chauffé à l’étuve. La substance retenue dans les eaux mères peut être récupérée en concentrant celles-ci au tiers de leur volume.
Le rendement est d’environ 13g de chlorhydrate de méthylamine.
1. Chauffer une prise d’essai avec une solution de soude caustique concentrée. Odeur, inflammabilité du gaz.
2. Une solution aqueuse concentrée du produit est additionnée de chlorure de platine : forme cristalline du chloroplatinate sous le microscope. À comparer avec le chloroplatinate d’ammonium.
3. Manière de se comporter de la solution aqueuse, acidulée, lorsqu’on l’additionne de quelques gouttes d’une solution de nitrite de soude et qu’on la chauffe doucement.
4. Réaction de l’isonitrile (voir aniline).
5. Réaction du sénévol. Dans un tube à essai, on introduit quelques cristaux de chlorhydrate de méthylamine, 1 à 2 gouttes de solution de soude caustique concentrée, une goutte de sulfure de carbone et 3 à 4 gouttes d’alcool. On ajoute alors un peu d’eau et un excès d’une solution diluée de nitrate d’argent ; on chauffe à l’ébullition, l’odeur très piquante du méthylsénévol se perçoit aussitôt.
La transformation d’un acide en aminé s’effectue également d’après la méthode de Curtius en passant par l’éthersel, l’hydrazide, l’azide et l’uréthane. (Journ. f. prakt. Chem. t. L, p. 275, 295 ; t. LII, p. 210 à 215, 227, 243).
28. Chlorure de benzyle C6H5.CH2Cl.
On introduit dans un ballon d’environ 300cm³ surmonté d’un réfrigérant à reflux, 100g de toluène et 5g de pentachlorure de phosphore qui agit comme chlorurant. On tare le ballon et son contenu et l’on porte à une douce ébullition en même temps qu’on fait passer un courant assez fort de chlore sec provenant d’une bombe.
L’opération est arrêtée lorsque l’augmentation du poids du ballon atteint à peu près 37g, ce qui correspond à l’absorption d’un atome de chlore. Cette augmentation a lieu très rapidement à la lumière du soleil, tandis que si le temps est couvert elle demande plusieurs heures.
La liqueur est soumise ensuite directement à la distillation fractionnée :
Il passe d’abord du toluène non transformé, puis du chlorure de benzyle qui constitue la fraction principale de 160° à 190° ; le résidu représente un mélange de produits plus riches en chlore (surtout du chlorure de benzal).
-
 Fig. 13.
Fig. 13. -
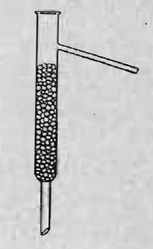 Fig. 14.
Fig. 14.


On purifie le chlorure de benzyle en le fractionnant de nouveau. On utilise pour cela non pas le ballon à distiller ordinaire, mais le dispositif de Linnemann (fig. 13) dans lequel les orifices inférieurs de chacune des 3 boules sont recouverts d’un treillis en platine, ou bien l’appareil à fractionnement de Henipel (fig. 14) rempli de perles de verre.
Point d’ébullition : 176°. Rendement : environ 50g.
29. Aldéhyde benzoïque C6H5.CHO.
Dans un ballon muni d’un réfrigérant ascendant, on chauffe pendant 5 à 8 heures 50g de chlorure de benzyle avec 40g de nitrate de cuivre et 250g d’eau jusqu’à ce qu’un échantillon de l’huile ne renferme plus du tout ou seulement des traces de chlore. Il est nécessaire, pour empêcher l’oxydation des vapeurs d’aldéhyde par l’air pendant cette longue opération, de faire passer dans l’appareil un lent courant de gaz carbonique. On empêchera également autant que possible l’accès de l’air dans les opérations ultérieures.
On extrait maintenant l’huile par de l’éther. Après évaporation de ce dernier, on secoue énergiquement l’huile avec une solution concentrée de bisulfite de soude (on emploie pour cela la solution commerciale à 40 pour 100 de NaHSO3.) La masse se prend alors en une bouillie cristalline représentant la combinaison bisulfitique de l’aldéhyde. Après quelques heures, on la filtre à la trompe et on la lave d’abord avec un peu d’eau, puis avec de l’alcool. Les cristaux, entièrement débarrassés de l’alcool de lavage au moyen d’une bonne filtration à la trompe, sont chauffés, avec de l’acide sulfurique dilué. L’aldéhyde, mise en liberté, est extraite par l’éther. La solution éthérée est desséchée sur du sulfate de soude calciné, l’éther évaporé et l’aldéhyde distillée. Point d’ébullition : 179°. Une perte notable est due à la facile oxydation de l’adéhyde benzoïque.
Rendement de 16g à 20g seulement.
30. Alcool benzylique C6H5.CH2.OH.
Dans un flacon bouché à l’émeri, d’une contenance de 300cm³ à 400cm³, on agite, jusqu’à émulsion persistante, 50g d’aldéhyde benzoïque et une solution froide de 45g de potasse solide dans 30g d’eau. On laisse le mélange au repos pendant 15 ou 20 heures. La masse devient solide grâce à la formation de benzoate de potasse. On ajoute de l’eau jusqu’à dissolution des cristaux, l’alcool benzylique se dissout également. C’est pourquoi l’on agite directement la liqueur à plusieurs reprises avec de l’éther et l’on traite la solution éthérée par du bisulfite de soude en solution pour éliminer l’aldéhyde benzoïque non transformée. On décante l’éther et on le sèche sur du sulfate de soude, puis on le filtre et on le distille ; l’huile qui reste est fractionnée. La plus grande partie passe dans l’intervalle de quelques degrés, c’est de l’alcool benzylique pur. Le rendement atteint environ les 90 pour 100 de la théorie. Point d’ébullition de l’alcool : 206°.
31. Benzoïne C6H5.CO.CHOH.C6H5.
Dans un ballon muni d’un réfrigérant ascendant, on chauffe pendant 15 à 20 minutes 50g d’aldéhyde benzoïque avec 5g de cyanure de potassium frais (produit commercial à 96 pour 100), 100g d’alcool et 100g d’eau. Par le refroidissement, la benzoïne se précipite sous la forme d’une bouillie cristalline. On l’essore à la trompe et cristallise le produit dans l’alcool chaud, après l’avoir décoloré par du charbon animal. On porte les premières eaux mères de nouveau à l’ébullition avec environ 2g de cyanure de potassium et la benzoïne formée est précipitée et traitée comme précédemment.
Rendement : à peu près 45g. Point de fusion : 134°.
32. Benzyle C6H5.CO.CO.C6H5.
On chauffe au bain-marie 20g de benzoïne avec de l’acide nitrique (poids spéc. 1,41), ce qui occasionne une vive réaction. Les cristaux se transforment bientôt en une huile jaune qui, au début, est un mélange de benzyle et de benzoïne. On a soin, en agitant fréquemment, de maintenir l’huile en contact intime avec l’acide.
L’oxydation dure d’une à deux heures. Le meilleur moyen de s’assurer si la transformation de la benzoïne est complète est un essai avec la liqueur de Fehling. À cet effet, on verse une goutte de l’huile dans de l’eau, où elle se prend en une masse cristalline, que l’on dissout dans de l’alcool. On étend d’eau, ajoute la liqueur de Fehling et chauffe entre 60° et 70°. Tant qu’il y a de la benzoïne, on observe une précipitation d’oxyde cuivreux. Dès que l’oxydation est terminée, on verse le produit dans l’eau et on filtre la masse cristalline qui s’est prise. On la dissout ensuite dans de l’alcool chaud. Par refroidissement, le benzyle se sépare en beaux prismes jaunes. Le rendement est très bon.
On contrôle la pureté du produit par la détermination du point de fusion : 90°.
33. Acide benzylique (C6H5)2:COH.C2H.
On fond dans un creuset d’argent ou de cuivre[7] 40g de potasse caustique avec un peu d’eau, puis on introduit dans la masse refroidie à 150°, et en remuant, 10g de benzyle sec. Ce dernier fond et se transforme bientôt en une masse solide de benzylate de potassium. Dès que toute l’huile a disparu, on laisse refroidir, on dissout le produit dans l’eau et l’on précipite l’acide benzylique en sursaturant par de l’acide chlorhydrique. On filtre après refroidissement et on lave le précipité cristallin avec de l’eau froide.
L’acide brut ainsi obtenu contient généralement de petites quantités d’acide benzoïque. Pour l’éliminer, on fait bouillir le mélange dans une capsule avec de l’eau, jusqu’à ce que l’odeur d’acide benzoïque ait disparu.
On purifie ensuite l’acide benzylique, non volatil, par recristallisation dans l’eau bouillante. Rendement presque quantitatif.
Point de fusion : 150°.
L’acide benzylique se dissout dans l’acide sulfurique concentré avec une magnifique coloration rouge violet, qui disparaît quand on dilue avec de l’eau.
34. Acide cinnamique C6H5.CH=CH.COOH.
Dans un ballon muni d’un réfrigérant à reflux et placé dans un bain d’huile, on chauffe à l’ébullition pendant 8 heures 20g de benzaldéhyde avec 30g d’anhydride acétique fraîchement distillé et 10g d’acétate de soude anhydre (c’est-à-dire fondu), finement pulvérisé. La masse encore chaude est versée dans 4 à 5 fois son poids d’eau ; on y fait alors passer un courant de vapeur d’eau jusqu’à ce que toute la benzaldéhyde qui n’a pas réagi soit entraînée. Simultanément, l’excès d’anhydride acétique se trouve éliminé ou bien transformé en acide acétique. La masse restante représente un mélange d’eau et d’acide acétique, avec une huile brune qui y est en suspension. Par refroidissement, il se précipite d’abondantes quantités d’acide cinnamique. Aussi traite-t-on immédiatement le mélange à chaud, sans le filtrer, par un excès de carbonate de soude solide. L’acide cinnamique se dissout transformé en sel de soude. Pour se débarrasser de l’huile insoluble, on filtre à chaud à travers un filtre à plis mouillé. En acidulant le filtrat avec de l’acide chlorhydrique, l’acide cinnamique se précipite déjà à chaud sous la forme d’une masse cristalline. Pour achever la cristallisation, on laisse refroidir, on filtre l’acide cinnamique précipité et on le purifie par une ou deux recristallisations dans l’eau chaude.
Détermination du point de fusion : 133°. Rendement : 15g à 20g.
35. Acide hydrocinnamique C6H5.CH2.CH2.CO2H.
Dans un flacon bouché à l’émeri, d’une contenance d’environ 200cm³ on dissout 10g d’acide cinnamique pulvérisé dans 50g d’eau et la quantité calculée de solution de soude caustique. On ajoute peu à peu à cette solution et en agitant fortement de l’amalgame de sodium à 2,5 pour 100[8]; 200g à 250g d’amalgame de bonne qualité suffisent.
Finalement, la solution alcaline est décantée du mercure et additionnée d’acide chlorhydrique. L’acide hydrocinnamique précipité est purifié par recristallisation dans la quantité suffisante d’eau. On contrôle la pureté du produit par la détermination du point de fusion 47°. Rendement très bon.
36. Hexahydrobenzène C6H12.
On humecte avec de l’eau un mélange à poids égaux d’oxyde de nickel et de pierre ponce pulvérisée comme du sable. Après dessiccation de la masse, on la réduit en poudre grossière et on l’introduit dans un tube à combustion long d’environ 40cm. On réduit alors au moyen d’hydrogène, en ayant soin de maintenir une température aussi basse que possible. La transformation se reconnaît au changement de coloration ; celle-ci passe du noir au gris clair. La réduction est terminée, lorsqu’il ne se dégage plus de vapeurs d’eau. On chauffe ensuite le tube de 180° à 190° dans un fourneau à pétrole de Volhard (fig. 15), tandis qu’un lent courant d’hydrogène sec passe, après avoir barboté dans un flacon contenant environ 30cm³ de benzène. L’hexahydrobenzène formé se condense dans le récipient refroidi par de la glace. Le courant d’hydrogène ne doit pas dépasser 100cm³ par minute. On peut, pour accélérer l’opération, chauffer le benzène à 30°. Après environ 7 heures, on interrompt l’opération, pèse le benzène non volatilisé et traite le distillat contenant encore un peu de benzène par un mélange nitrant d’acides sulfurique et nitrique. L’huile, plus légère, est séparée de l’acide, lavée avec de l’eau et séchée sur du chlorure de calcium. L’hexahydrobenzène passe au fractionnement entre 80° et 82°.

Rendement : au moins 80 pour 100 du benzène volatilisé.
37. Éther acétylacétique CH3.CO.CH2.CO2.C2H5.
On introduit 30g de sodium, étiré en fils à l’aide de la presse à sodium, dans un ballon contenant 300g d’éther acétique commercial, de la meilleure qualité. Le ballon est immédiatement relié à un réfrigérant ascendant. Le liquide commence bientôt à bouillir. Dès que le dégagement de chaleur diminue, on chauffe au bain-marie jusqu’à dissolution complète du métal. La masse liquide, encore chaude et bien agitée, est additionnée avec précaution d’acide sulfurique dilué (1:5) jusqu’à réaction acide. On laisse refroidir. Quand les deux liquides, bien mélangés par l’agitation, se sont de nouveau séparés, on décante la couche supérieure et on la lave encore une fois avec un peu d’eau. En distillant ensuite au bain-marie, on chasse la majeure partie de l’éther acétique non transformé. Le résidu est soumis plusieurs fois à la distillation fractionnée. La fraction 175° à 185° renferme de l’éther acétylacétique presque pur. Le résidu jaune qui se trouve dans la cornue se prend par refroidissement en une bouillie cristalline d’acide déhydracétique. Le rendement en éther acétylacétique s’élève au plus à 50g, mais, souvent, lorsque la dissolution du métal dure longtemps, ou, ce qui revient au même, si l’on opère en une fois avec de plus grandes quantités, il demeure bien en dessous de ce chiffre.
1. Coloration avec le chlorure ferrique.
2. Solubilité dans les alcalis. Les acides précipitent de nouveau l’éther. Si, par contre, on chauffe quelque temps la solution alcaline, l’éther est complètement décomposé et fournit de l’alcool, de l’acide carbonique, de l’acétone et de petites quantités d’acide acétique. Il subit une décomposition semblable lorsqu’on le chauffe longtemps avec des acides dilués.
38. Éther de l’acide diacetsuccinique
| CH3.CO.CH | CH.CO.CH3 | |
| — | — | |
| C2H5.O2C | CO2.C2H5 |
Dans un flacon d’environ 500cm³ muni d’un réfrigérant ascendant, on dissout 15g d’éther acétylacétique dans 150g d’éther pur, séché sur du sodium et l’on ajoute à cette solution 5g de sodium, de préférence sous la forme de fils minces, que l’on prépare à l’aide d’une presse. Dans cette opération, on reçoit le fil directement dans de l’éther pur ou, ce qui est préférable, dans du toluène, afin de le préserver de l’humidité de l’air. Il se produit un vif dégagement d’hydrogène et le sodium se transforme peu à peu en une masse très finement divisée d’éther acétacétique sodé. La réaction principale est terminée au bout de 2 à 3 heures. Une partie du sodium cependant est enveloppée de telle sorte par l’éther sodé qu’il n’est plus attaqué. On bouche alors le flacon pour un instant et l’on agite fortement. La surface du métal réapparaît brillante, et le dégagement d’hydrogène recommence. En répétant plusieurs fois cette opération, le métal disparaît en un temps relativement court.
Ceci fait, on dissout 20g d’iode, finement pulvérisé, dans de l’éther pur et l’on verse cette solution, par petites portions et en agitant constamment, dans l’éther acétacétique sodé. La réaction se produit immédiatement et il se dépose une quantité considérable d’iodure de sodium. Aussitôt que la couleur de l’iode ne disparaît plus immédiatement après son addition, on filtre la solution et l’on évapore l’éther. Le résidu est l’éther diacetsuccinique que l’on presse sur une plaque poreuse, après qu’il s’est pris par refroidissement. On le recristallise dans de l’acide acétique chaud à 50 pour 100.
(L. Knorr, Ber. d. d. chem. Ges., t. XIX, p. 46.)
On dissout une petite quantité d’éther diacetsuccinique dans de l’acide acétique glacial. On ajoute une solution d’ammoniaque dans un excès d’acide acétique et l’on fait bouillir le mélange pendant environ une demi-minute ; on verse ensuite de l’acide sulfurique dilué et l’on porte de nouveau à l’ébullition en introduisant une bûchette de bois de pin dans la solution. Une coloration rouge intense du bois indique la formation d’un dérivé pyrrolique.
39. Malonate diéthylique CH2(CO2C2H5)2.
100g d’acide chloracétique broyés sont additionnés de 150g de glace concassée et dissous dans 125g de solution de soude caustique à 33 ⅓ pour 100. Si la liqueur est encore acide, on la neutralise exactement avec de la soude caustique. Puis on ajoute une solution de 69g de cyanure de potassium dans 130g d’eau, solution qui doit avoir une température de 40°. Le mélange s’échauffe de lui-même et la température s’élève jusqu’à 50° ou 60°. Au bout d’une heure, on chauffe lentement à 100° et maintient cette température pendant une heure. Puis on laisse refroidir à 20°, ajoute encore 125g de soude caustique de la concentration indiquée et chauffe de nouveau à 100°, jusqu’à ce qu’il ne se dégage plus d’ammoniaque, ce qui est le cas d’ordinaire après 2 ou 3 heures. La transformation de l’acide cyanacétique en acide malonique est achevée, quand une prise d’essai de la liqueur, additionnée de soude caustique, ne donne plus d’ammoniaque à l’ébullition. On ajoute alors une solution chaude de chlorure de calcium, à 25 pour 100 environ, jusqu’à ce qu’il ne se forme plus de précipité ; il faut pour cela 120g à peu près de chlorure de calcium commercial sec ou la quantité correspondante du sel cristallisé. Le malonate de calcium précipité constitue au début une bouillie épaisse, amorphe, qui se transforme complètement au bout de 24 heures en une masse cristalline. On l’essore à la trompe, on la lave avec très peu d’eau froide et on l’exprime ensuite à la presse. Pour finir, on sèche le sel au bain-marie ; après ce traitement, il renferme encore un peu d’eau de cristallisation, qui part à 100° dans le vide, mais dont l’élimination n’est pas nécessaire pour la préparation suivante. Le rendement s’élève aux 90 à 95 pour 100 de la théorie.
Pour effectuer l’éthérification à partir de l’alcool et de l’acide chlorhydrique gazeux, on prend, pour 100g de sel de calcium séché soigneusement au bain-marie, 250g d’alcool absolu. L’opération se fait dans un ballon d’une contenance de 1l. Comme le malonate de calcium se rassemble facilement en une masse compacte et qu’il est alors difficilement attaqué, il convient d’abord d’en prendre 20g environ et de les recouvrir avec la quantité totale de l’alcool, puis de faire passer un fort courant d’acide chlorhydrique bien sec. Au fur et à mesure que le sel disparaît, on en ajoute de nouvelles portions ; chaque fois 15g à 20g dans l’intervalle d’une demi-heure, jusqu’à ce que tout soit dissous et que la solution alcoolique soit saturée par l’acide chlorhydrique. Après 24 heures de repos, on neutralise la solution par addition de carbonate de calcium en poudre (craie) et distille l’alcool sous pression réduite. L’éther qui reste est extrait du liquide aqueux au moyen d’éther. On sèche la solution éthérée avec du chlorure de calcium et, après distillation, on fractionne le résidu.
Point d’ébullition : 197° à 198° (corr.). Rendement : 75 pour 100 de la théorie.
40. Éther benzylmalonique C6H5.CH2.CH(CO2C2H5)2.
On dissout 7g,2 de sodium (1at) dans 150cm³ d’alcool absolu, puis on ajoute 50g de malonate d’éthyle (1mol) et 43g de chlorure de benzyle pur commercial (1mol). Au bout de quelques minutes, la solution claire s’échauffe fortement, tandis qu’il y a précipitation de chlorure de sodium ; après 10 minutes, on évapore l’alcool au bain-marie, sans l’avoir préalablement filtré. Vers la fin, la réaction de la liqueur n’est plus que faiblement alcaline. On additionne le résidu d’eau, afin de dissoudre le chlorure de sodium, et l’on extrait par l’éther l’huile qui s’est séparée. La solution éthérée est séchée avec du carbonate de potasse ou du sulfate de soude calciné, l’éther évaporé et le résidu fractionné sous pression très réduite. Sous une pression de 11mm, la plus grande partie de l’éther benzylmalonique passe entre 166° et 169° en donnant une huile incolore et fluide. On recueille le distillat jusqu’à 175°. Le rendement est de 45g à 50g (calculé 78g). Un nouveau fractionnement donne 40g à 45g d’un produit bouillant de 166° à 168°.
L’éther bout à 173° sous 15mm de pression, à 183° sous 20mm, et à 188° sous 25mm.
41. Acide benzylmalonique C6H5.CH2.CH(COOOH)2.
Dans un ballon à fond rond d’environ 250cm³ on émulsionne 30g d’éther benzylmalonique en les agitant fortement avec 35cm³ de solution de potasse caustique concentrée (p. spec, 1,32) (2mol,2). La dissolution se fait tout d’un coup, si l’on chauffe doucement au bain-marie. En chauffant encore pendant 1 heure, on achève la saponification, tout en chassant la plus grande partie de l’alcool formé ; une petite quantité d’huile se sépare à ce moment ; on l’élimine par agitation avec de l’éther. On ajoute ensuite à la solution aqueuse un peu plus d’acide chlorhydrique dilué que la quantité équivalente à l’alcali employé. Pour extraire l’acide benzylmalonique mis en liberté, on agite la solution aqueuse d’abord avec 50cm³, puis, à plusieurs reprises, chaque fois avec 25cm³ d’éther.
La solution éthérée, après avoir été desséchée sur du sulfate de soude calciné, est filtrée, et l’éther est complètement évaporé dans le dessiccateur à vide. Le résidu, à peine coloré, se solidifie entièrement. Rendement, 21g. Pour recristalliser le produit brut, on le dissout dans 140cm³ de benzène chaud. Point de fusion : 117° (corr.). Rendement en produit pur : 19g,5 (théorie : 24g).
Dans un tube à essai à large ouverture, on chauffe 1g d’acide benzylmalonique à 180°, en se servant d’un bain. Il se produit un vif dégagement d’anhydride carbonique, qui cesse au bout de 10 minutes environ. La masse se prend par refroidissement. On la recristallise dans l’eau chaude ; elle présente alors le point de fusion de l’acide hydrocinnamique : 47°. Rendement presque quantitatif.
42. Acide téréphtalique C6H4(CO2H)2.
Dans un ballon muni d’un réfrigérant ascendant, on chauffe 10g de p-xylène commercial avec un mélange de 40g du bichromate de potasse, 30cm³ d’acide sulfurique concentré et 6cm³ d’eau. La solution se colore peu à peu en vert, et l’acide téréphtalique se dépose, surtout contre les parois du ballon, sous la forme d’une masse légère, peu colorée. Au bout de 15 heures, on chasse par les vapeurs d’eau l’hydrocarbure non transformé, et l’on filtre la solution après refroidissement. Pour purifier l’acide téréphtalique brut, on le dissout dans une solution diluée et chaude de carbonate de soude et on le reprécipite du filtrat par de l’acide chlorhydrique. Rendement : 4g. La caractéristique de cet acide est sa très faible solubilité dans l’eau, l’alcool et l’éther.
43. Acide pyruvique CH3.CO.CO2H.
100g d’acide tartrique et 240g de pyrosulfate de potasse finement pulvérisés sont soigneusement mélangés et introduits dans un ballon à fond rond, d’une contenance d’au moins 2l et dont le col est aussi court que possible. Le ballon est placé dans un bain d’huile dont la température ne doit pas dépasser 220° ; le mélange distille. Pendant la distillation, dont la durée est d’environ 4 à 5 heures, une grande quantité de gaz fortement odorants se dégage (on utilise la hotte) et il passe une solution aqueuse d’acide pyruvique. On interrompt la distillation quand il ne se condense plus de gouttelettes huileuses en quantité notable dans le réfrigérant. On soumet immédiatement le distillat à la distillation fractionnée. La fraction passant entre 130° et 180° est recueillie à part et purifiée par une nouvelle rectification. L’acide pyruvique purifié bout de 165° à 170°. Rendement : 20g à 25g.
Si l’on mélange une solution acétique de phénylhydrazine avec une solution aqueuse d’acide pyruvique, le produit de la réaction se sépare aussitôt en belles aiguilles jaunes. Il sert à caractériser l’acide pyruvique.
44. Épichlorhydrine CH2Cl-CHO-CH2.
Dans une capsule et sous la hotte, on chauffe 200g de glycérine jusqu’à ce que le thermomètre marque 170°, de façon à chasser l’eau ; puis on dissout la glycérine ainsi séchée dans un volume égal d’acide acétique glacial. On sature cette solution, à la température ordinaire, par un fort courant d’acide chlorhydrique gazeux obtenu et séché de la façon habituelle. On chauffe ensuite pendant environ 6 heures au bain-marie, tout en continuant à faire passer le gaz chlorhydrique. Après un repos de 12 heures, le liquide est soumis à la distillation fractionnée. Il se dégage d’abord une grande quantité d’acide chlorhydrique, puis il passe de l’acide acétique aqueux et, finalement, un mélange de dichlorhydrine et d’acétodichlorhydrine. La fraction, qui passe de 160° à 220°, est recueillie à part et transformée en épichlorhydrine, sans autre purification. On peut encore retirer, de la fraction de 110° à 160°, une petite quantité du même produit, à l’état huileux, en l’additionnant d’eau. Le rendement total en produit brut s’élève à peu près à 120 pour 100 de la glycérine employée.
Pour transformer le produit en épichlorhydrine, on le traite par de la potasse aqueuse. Dans ce but, on dissout 100g de potasse caustique dans deux fois son poids d’eau. Lorsque cette solution a pris la température de la chambre, on la verse peu à peu dans la dichlorhydrine brute, en remuant constamment et en refroidissant avec de l’eau. Le produit huileux se transforme en épichlorhydrine, liqueur facilement mobile. La transformation s’accomplit aisément à la température ordinaire et presque complètement ; il faut toutefois éviter soigneusement tout échauffement un peu fort de la solution alcaline, autrement l’épichlorhydrine serait à son tour saponifiée. L’opération terminée, l’épichlorhydrine est extraite à plusieurs reprises par l’éther et la solution desséchée sur du sulfate de soude. On filtre, on évapore l’éther et l’on fractionne le résidu (voir le chlorure de benzyle). La fraction passant au-dessus de 130° est, dans sa majeure partie, de l’acétodichlorhydrine non transformée ; on la traite encore une fois par de la lessive de potasse.
Point d’ébullition de l’épichlorhydrine : 119°. Rendement : environ 45g.
L’épichlorhydrine se dissout dans une solution chaude de potasse caustique, en se transformant en glycérine.
45. Acroléine CH2:CH.COH.
200g de glycérine desséchée d’après la méthode indiquée au n° 44 sont introduits dans un ballon à fond rond ou de préférence dans un récipient de métal de la forme ci-contre (fig. 16). On ajoute 400g de bisulfite de potassium, concassé en petits morceaux de la grosseur d’une lentille. Il est avantageux de boucher le ballon ou la cornue métallique et de conserver le mélange plusieurs jours à la température ordinaire, avant de commencer l’opération.
a est un récipient cylindrique en cuivre ou de préférence en fer, terminé par un fort rebord. Sur celui-ci, on peut fixer un couvercle b au moyen des vis c. Pour que la fermeture soit hermétique, on mettra un anneau de papier d’amiante entre le couvercle et le rebord. Par l’orifice du couvercle passe un tube de verre recourbé d aboutissant à un réfrigérant. Celui-ci est relié, au moyen d’un raccord hermétique, à un ballon à distiller ou de préférence à un récipient en fer-blanc à deux tubulures. Ce récipient est placé au milieu d’un mélange réfrigérant et possède un tube de dégagement, aboutissant dans une cheminée à fort tirage.

Le mélange de glycérine et de bisulfite est chauffé lentement sur un fourneau à gaz. Au commencement, il ne distille presque rien que de l’eau ; puis le mélange brunit et augmente de volume. Il passe alors, à côté de l’eau et de l’acide sulfureux, une quantité assez considérable d’acroléine. La distillation dure plusieurs heures. On l’interrompt, lorsqu’il ne passe pour ainsi dire plus de liquide.
Le liquide distillé forme deux couches, dont la supérieure est de l’acroléine et l’inférieure de l’eau. Celle-ci contient une grande quantité d’anhydride sulfureux. Pour s’en débarrasser, on ajoute au mélange, par petites portions, de l’oxyde de plomb pulvérisé, jusqu’à ce que celui-ci ne se transforme plus en sulfite de plomb blanc (après chaque addition d’oxyde de plomb, il faut secouer énergiquement). Ceci fait, toute la masse est distillée au bain-marie ; le liquide qui passe est recueilli dans un récipient soigneusement refroidi.
À cause de la mauvaise odeur de l’acroléine, toutes ces opérations doivent se faire sous la hotte. Le produit obtenu est séché par du chlorure de calcium et distillé encore une fois au bain-marie. Plus rapidement sera exécutée sa préparation, plus petite sera la perte par polymérisation. Le rendement est de 35g environ.
Si l’on garde l’acroléine, elle finit par se polymériser. Cette polymérisation s’effectue dans l’espace de quelques minutes, si l’on ajoute à l’acroléine un peu d’alcali ou une solution de cyanure de potassium.
46. Ortho- et para-nitrophénol C6H4 (NO2)(OH).
Dans 300 g d’acide nitrique (d = 1,11) bien refroidi par un courant d’eau, on introduit par petites portions 50g de phénol cristallisé, en agitant après chaque addition. Le mélange se colore dès le commencement en brun foncé ; au bout de peu de temps une huile épaisse et foncée se dépose. On laisse la réaction se poursuivre pendant quelques heures à froid en ayant soin d’agiter de temps en temps. On sépare la couche acide de la couche huileuse par décantation ; on lave cette dernière plusieurs fois avec de l’eau et on la soumet à la distillation avec les vapeurs d’eau. L’orthonitrophénol seul est entraîné par les vapeurs, sous forme d’une huile jaune clair, qui se solidifie en longues aiguilles dans le récipient. Rendement : 15g à 20g. L’orthonitrophénol obtenu est pur. Si l’on veut l’obtenir en beaux cristaux, on procède de la façon suivante : le produit obtenu par distillation avec les vapeurs d’eau est soigneusement séché entre deux feuilles de papier à filtrer. On le dissout dans de l’éther absolu et l’on ajoute à cette solution environ 1 ½ fois son volume d’éther de pétrole. Par simple évaporation à l’air, il y a dépôt de superbes cristaux d’orthonitrophénol qui fondent à 45°.
Le résidu résineux, non entraîné par les vapeurs d’eau, contient le paranitrophénol. On le traite à chaud par de la soude caustique diluée et du noir animal et l’on filtre. La solution alcaline brun foncé est évaporée et additionnée de soude caustique très concentrée (1 : 1). Par refroidissement, le sel de soude du paranitrophénol se dépose en une masse cristalline jaune. Ces cristaux sont étendus sur une assiette poreuse pour être débarrassés de leur eau mère, puis dissous dans peu d’eau. Par addition de soude caustique, le sel de soude se dépose. Celui-ci, traité par les acides, donne une huile qui ne tarde pas à cristalliser. C’est le paranitrophénol, qu’on dissout dans l’eau chaude ; par addition de quelques gouttes d’acide chlorhydrique dilué, on obtient une belle cristallisation. Point de fusion : 114°.
47. Acide picrique C6H2(NO2)2.OH.
On mélange 10g de phénol avec 10g d’acide sulfurique concentré. On introduit cette masse, par très petites portions, dans 30g environ d’acide nitrique (d = 1,4). Il se produit une vive réaction et un fort dégagement de vapeurs rouges. Le mélange est chauffé 1 ou 2 heures au bain-marie, c’est-à-dire aussi longtemps que l’acide nitrique réagit encore. Le liquide perd la teinte foncée qu’il avait au commencement et prend une coloration jaune d’or. En même temps, il se sépare souvent une huile jaune foncé qui consiste, pour la plus grande partie, en dinitrophénol. Pour transformer quantitativement ce dernier en acide picrique, on ajoute avec précaution au mélange de l’acide nitrique concentré et l’on chauffe encore au bain-marie. On répète cette opération jusqu’à ce qu’on obtienne un liquide brun clair et qu’une petite portion de la solution, diluée avec de l’eau, abandonne des cristaux jaunes. Ces cristaux doivent se dissoudre dans l’eau bouillante en donnant une solution parfaitement claire, sans résidu huileux. Après s’être assuré de cette façon que la transformation était complète, on verse tout le liquide dans de l’eau. Par refroidissement, l’acide picrique cristallise. On le filtre et on le recristallise de l’eau chaude.
48. Anisol C6H5.OCH3.
Dans un ballon à fond rond, d’une capacité de 500cm³, on dissout 30g de phénol dans 100cm³ d’eau, additionnés de 40cm³ d’une solution dix fois normale de soude caustique. On verse dans le mélange 5cm³ de sulfate de méthyle du commerce[9] et l’on secoue énergiquement. Le commencement de la réaction se manifeste par le fait que le liquide devient aussitôt d’un blanc laiteux. Le sulfate de méthyle, qui s’était rassemblé au fond du gallon, disparaît, tandis qu’une couche d’huile se forme au-dessus de la solution aqueuse. En même temps, la masse s’échauffe assez fortement. En plongeant le ballon dans l’eau froide, il faut s’arranger pour que la température se maintienne entre 40° et 50° (contrôler cette température par un thermomètre plongeant dans le liquide). La réaction est terminée dès que la solution ne s’échauffe plus et que la température tombe au-dessous de 40°, sans qu’on ait besoin de refroidir le ballon. Pour détruire l’excès de sulfate de méthyle, on fait bouillir pendant quelques minutes, en secouant constamment. Après refroidissement, on ajoute de la soude caustique, jusqu’à réaction alcaline. On verse le liquide dans un entonnoir à robinet et l’on secoue énergiquement avec de l’éther. Après avoir laissé reposer, on sépare la couche aqueuse de la couche éthérée. On sèche cette dernière sur du carbonate de potassium desséché au rouge (le plus simple est de secouer dans l’entonnoir à décantation lui-même la solution éthérée avec le carbonate). Après filtration, on distille l’éther au bain-marie. Le résidu est purifié par distillation fractionnée ; l’anisol bout à 155°. Le rendement atteint les 90 pour 100 du rendement théorique.
49. Quinone et Hydroquinone C6H4O2 et C6H6O2.
On dissout 29g d’aniline dans 600cm³ d’eau et 160g d’acide sulfurique. On refroidit la solution à 10°-12°. On maintient cette température pendant qu’on ajoute, dans l’espace d’une heure et en agitant continuellement, 20g de bichromate de potassium en poudre, par portions de 1g. On laisse reposer pendant la nuit. On introduit de nouveau, de la même façon et à la même température, 40g de bichromate, jusqu’à ce que la coloration ait passé du bleu foncé au brun. On secoue plusieurs fois la solution avec d’assez grandes quantités d’éther. Si le liquide donne une émulsion, on ajoute à l’éther quelques centimètres cubes d’alcool qui font tomber les petites particules solides en suspension. Après séparation des deux couches et distillation de l’éther, la quinone reste sous forme d’une masse cristalline jaune brun. On obtient environ 20g de quinone brute. On purifie généralement la quinone par distillation avec les vapeurs d’eau et cela de la façon suivante : on introduit le produit brut desséché dans un ballon à distiller, au travers duquel on fait passer un violent courant de vapeurs d’eau. Celui-ci entraîne rapidement la quinone, qui se prend en cristaux jaune clair dans le réfrigérant et le récipient. Par ce procédé, on ne perd que peu de substance : 20g de quinone donnent environ 17g d’un produit chimiquement pur.
Pour obtenir l’hydroquinone, on pulvérise la quinone aussi finement que possible et on la met en suspension dans l’eau. On verse dans ce mélange une solution aqueuse d’anhydride sulfureux[10] jusqu’à complète dissolution et décoloration. On épuise plusieurs fois la solution par l’éther. Par distillation de ce dernier, l’hydroquinone reste en une masse cristalline incolore. Dans la réduction de la quinone en hydroquinone, il faut éviter la formation de la quinhydrone qui est vert foncé.
50. Aldéhyde salicylique C6H4(COH)OH.
On introduit dans un ballon, muni d’un réfrigérant ascendant, 50g de phénol et une solution de 100g de soude caustique dans 110g d’eau, puis on chauffe à 50°-60°. Par le tube du réfrigérant on verse, par très petites portions à la fois, 75g de chloroforme. Une réaction énergique se produit. La solution, qui était d’abord légèrement jaune, se colore en violet, puis en rouge foncé ; lorsque tout le chloroforme a été ajouté, on chauffe encore une demi-heure au réfrigérant ascendant ; puis on distille l’excès de chloroforme. On acidule la solution aqueuse par de l’acide sulfurique dilué et l’on distille avec les vapeurs d’eau, jusqu’à ce que le liquide qui passe n’entraîne plus de gouttes d’huile. Le produit de distillation, qui contient du phénol et de l’aldéhyde salicylique, est agité avec de l’éther. On concentre légèrement la solution éthérée et on la secoue énergiquement et assez longtemps avec une solution concentrée de bisulfite de soude.
La combinaison de l’aldéhyde salicylique avec le bisulfite se dépose sous forme de fines paillettes brillantes qui donnent avec le liquide une bouillie cristalline. L’éther retient le phénol. Lorsque la solution éthérée, secouée avec le bisulfite de soude, ne donne plus de cristaux, on filtre à la trompe et on lave à l’alcool pour entraîner tout l’éther qui contient encore du phénol. Les cristaux sont chauffés au bain-marie avec un peu d’acide sulfurique dilué. Après refroidissement, on extrait l’aldéhyde par l’éther, qu’on distille ensuite. On sèche l’aldéhyde sur du chlorure de calcium, puis on le distille ; il bout à 196°.
Le rendement atteint environ les 17 pour 100 du phénol employé.
51. Acide β-naphtaline sulfonique C10H7.SO3H.
On chauffe, à 90°-100°, 120g d’acide sulfurique à 96 pour 100 ; on y ajoute peu à peu, en agitant continuellement, 100g de naphtaline finement pulvérisée. Lorsque tout a été ajouté (ce qui prend 15 minutes environ), on élève lentement la température jusqu’à 160° et on la maintient ainsi pendant 12 heures. De la naphtaline pure doit être presque toute sulfonée au bout de ce temps et le produit de réaction doit se dissoudre dans l’eau, en ne donnant qu’un très léger trouble. On verse la solution dans 1l,5 d’eau, et l’on neutralise à l’ébullition par du lait de chaux. On filtre à chaud sur de la toile, on presse bien le sulfate de chaux qui s’était formé et qu’on fait bouillir encore une fois avec 1l d’eau ; on le filtre et on le presse de nouveau comme tout à l’heure. On réunit les deux liquides et on les concentre jusqu’à ce qu’une prise d’essai donne, par refroidissement, une pâte épaisse.
On laisse reposer 24 heures, puis on filtre et l’on presse fortement le sel de chaux qui a cristallisé. Les eaux mères contiennent les sels de l’acide α-monosulfonique et de l’acide disulfonique, qui sont plus facilement solubles.
On transforme ensuite le β-naphtalino-sulfonate de chaux en sel de soude. Pour cela, on dissout le sel de chaux dans beaucoup d’eau bouillante et l’on ajoute à l’ébullition du carbonate de soude, jusqu’à ce qu’un essai filtré ne donne plus de précipité avec ce réactif.
La solution du sel de soude est filtrée à l’étamine et concentrée jusqu’à commencement de cristallisation à chaud. On laisse cristalliser par refroidissement ; on presse et l’on sèche le sel humide au bain-marie.
Si l’on concentre les eaux mères du sel de soude, on peut récupérer encore quelques grammes du produit. En opérant bien, on obtient, en partant de 100g de naphtaline, 130g à 150g de β-naphtaline-sulfonate de soude exempt d’acide α-sulfonique et d’acide disulfoné.
52. β-naphtol C10H7.OH.
On introduit dans un creuset de cuivre 300g de soude aussi pure que possible et 30cm³ d’eau. On chauffe avec une forte flamme jusqu’à 280°, en agitant continuellement avec une spatule en cuivre. (Il faut avoir bien soin de protéger ses yeux et ses mains. La température doit être prise au moyen d’un thermomètre enveloppé d’une douille de cuivre.) On ajoute, aussi vite que possible et toujours en remuant, 100g de β-naphtaline-sulfonate de soude sec très finement pulvérisé. L’addition d’une nouvelle portion se règle d’après la température qui ne doit jamais descendre au-dessous de 260°. Lorsque tout le sulfonate a été ajouté, on élève rapidement la température jusqu’à 320°, en agitant toujours.
L’opération se passe de la façon suivante :
La soude, qui était tout à fait liquide à 280°, s’épaissit par addition du β-naphtaline-sulfonate de soude. Le mélange se laisse cependant bien remuer si la température ne tombe pas au-dessous de 260°. Lorsque tout le sel a été introduit et que la température a atteint 300°, la masse augmente de volume, par ébullition de l’eau, et se transforme en un liquide visqueux jaune clair. Ce phénomène caractéristique indique le commencement de la réaction qui, entre 310° et 320°, est terminée en quelques minutes. Il y a un vif dégagement de vapeurs d’eau et formation de mousse et de bulles d’air.
On reconnaît la fin de la réaction à ce que le produit, de jaune et visqueux qu’il était, devient foncé et tout à fait liquide, et à la formation de deux couches apparaissant dès que l’on cesse de remuer. La partie supérieure est jaune brun, claire et transparente et se compose pour la plus grande partie de naphtol sodé, avec un peu de soude caustique et de sulfite. Après séparation du mélange en deux couches, on éloigne la flamme et l’on décante le naphtol sodé, ou bien, après refroidissement, on le sépare mécaniquement de la couche inférieure de soude caustique. On dissout le naphtol sodé dans l’eau bouillante et on le traite à chaud par de l’acide chlorhydrique à 15 pour 100. Le β-naphtol se dépose. Après refroidissement, on le filtre à la trompe et on le lave à l’eau. On le cristallise de l’eau bouillante.
En partant de 100g de β-naphtaline-sulfonate de soude pur, on obtient 55g de β-naphtol.
53. Réduction du naphtol en naphtaline.
Cette opération présente dans son exécution une grande analogie avec une détermination d’azote. L’oxyde de cuivre est remplacé par de la poudre de zinc. Afin que celle-ci présente une grande surface, on procède ainsi : de tout petits morceaux de pierre ponce sont humectés d’eau et roulés dans de la poudre de zinc jusqu’à ce que toute leur surface en soit imprégnée. On les fait sécher pendant 4 heures à 150°. Le tube à combustion est étiré à l’extrémité par laquelle arrivera un courant d’hydrogène. On remplit le tube de la façon suivante : on met d’abord, sur une longueur de 10cm, de la pierre ponce imbibée de zinc, puis un mélange intime de 3g de β-naphtol et de 30g de poudre de zinc, et enfin, jusqu’aux deux tiers du tube, de la pierre ponce recouverte de zinc. En frappant légèrement le tube, on ménage un canal pour le passage des gaz et des vapeurs. Le tube, ainsi préparé, est placé dans un fourneau à combustion légèrement incliné. La partie ouverte du tube doit être la plus basse, et toute la partie vide doit se trouver en dehors du fourneau. On commence par diriger un courant d’hydrogène sec à travers le tube. Lorsque tout l’air a été chassé, on modère le courant et l’on chauffe la pierre ponce jusqu’au rouge sombre. On chauffe enfin le mélange de β-naphtol et de zinc. La naphtaline formée distille et sublime dans la partie vide du tube, située en dehors du fourneau. Si par hasard le tube se bouche par amas de substance sublimée, on l’introduit un peu plus avant dans le fourneau, et l’on chauffe jusqu’à ce que la masse ait fondu et sublimé un peu plus loin. Le courant d’hydrogène n’entraîne au dehors que de très petites quantités de naphtaline. Si l’on veut récupérer ces dernières, on munira le tube d’une rallonge conduisant les vapeurs dans un appareil réfrigérant. On agira de même dans tous les autres cas semblables où l’on aura affaire à des produits de distillation facilement volatils.
La distillation dure 45 minutes. Lorsqu’elle est terminée, on casse la partie du tube contenant la naphtaline et l’on recueille l’hydrocarbure avec une spatule. On le dissout dans l’alcool chaud et on le fait bouillir un moment avec du noir animal. On filtre et on laisse cristalliser. Si le produit est encore coloré, on le purifie complètement par sublimation. La naphtaline fond à 79°. Rendement 1g.
54. Cyanate de potassium et urée.
KOCN et NH2.CO.NH2.
On prend du ferrocyanure de potassium du commerce qu’on casse on petits morceaux et qu’on étend en couche mince dans une grande capsule en fer. On chauffe modérément, jusqu’à ce que les cristaux aient perdu leur éclat ; on les pulvérise à chaud aussi finement que possible. On étend de nouveau le ferrocyanure pulvérisé sur la même capsule et on le dessèche encore une fois de la même façon pendant quelques heures. Il faut s’assurer, en chauffant la poudre dans un tube à essais, qu’elle ne contient plus trace d’eau.
On prend 200g de ce ferrocyanure de potassium complètement desséché, qu’on mélange intimement et à chaud avec 150g de bichromate de potassium fondu. On introduit ce mélange par petites portions de 5g à 6g dans la capsule déjà employée, qu’on chauffe au moyen d’une très forte couronne de gaz. Des taches noires apparaissent au milieu du mélange et l’envahissent peu à peu ; on agite continuellement la masse au moyen d’une spatule en fer. Il ne doit pas se dégager d’ammoniaque. Le produit de réaction, devenu tout à fait noir, est broyé à chaud, mis dans un ballon muni d’un réfrigérant, et placé sur le bain-marie. On le traite pendant 2 minutes, tout en secouant fortement, par un mélange déjà chaud de 900cm³ d’alcool à 80 pour 100 et de 100cm³ d’alcool méthylique. On décante la solution sur un filtre à plis et l’on reçoit le liquide limpide dans un Erlenmeyer refroidi par de la glace. Le cyanate cristallise. Les eaux mères alcooliques sont remises dans le ballon contenant la masse noire ; on fait bouillir 2 minutes et l’on décante comme auparavant. On recommence l’épuisement plusieurs fois de cette manière ; cinq ou six fois suffisent généralement. On essore sur le même filtre les différentes cristallisations de cyanate obtenues, on les lave à l’éther et on les sèche sur de l’acide sulfurique. Rendement : 80g à 90g.
Les eaux mères alcooliques, qui contiennent encore du cyanate, peuvent être utilisées pour la préparation de l’urée. Pour cela, on ajoute à ces eaux mères, tout en les remuant, une solution de 70g de sulfate d’ammonium dans 100cm³ d’eau chaude. On filtre, pour se débarrasser du sulfate de potassium formé ; on distille l’alcool, on évapore à sec et l’on extrait l’urée du résidu au moyen d’alcool à 96 pour 100. En recristallisant l’urée de l’alcool amylique bouillant (hotte !), on l’obtient tout à fait pure.
1. Précipitation par l’acide nitrique.
2. Précipitation par le nitrate mercurique.
3. Décomposition par les alcalis bouillants, avec dégagement d’ammoniaque.
4. Décomposition par l’acide nitreux, même à froid, avec dégagement d’azote et d’anhydride carbonique.
55. Alloxane et alloxantine.
On introduit dans un ballon 15g d’acide urique[11] finement pulvérisé, 30g d’acide chlorhydrique concentré (d = 1,19) et 40g d’eau. On chauffe le mélange à 30° et l’on y ajoute par petites portions 4g de chlorate de potassium en poudre ; cette addition doit durer 45 minutes. L’acide urique, à part quelques impuretés, se dissout entièrement. On filtre à chaud et l’on dilue avec 30cm³ d’eau, puis on sature la liqueur, à température ordinaire, par un assez fort courant d’hydrogène sulfuré. Il y a d’abord dépôt de soufre amorphe, bientôt accompagné de cristaux d’alloxantine. Cette cristallisation peut être activée au moyen d’un bon refroidissement. On filtre et on lave le précipité avec de l’eau froide ; puis on le reprend par de l’eau bouillante. On filtre pour se débarrasser du soufre, resté insoluble. Par refroidissement, l’alloxantine se dépose pure en beaux prismes blancs. Rendement : 10g à 12g.
1. L’eau de baryte donne une coloration avec les solutions d’alloxantine.
2. La solution d’alloxantine réduit la liqueur ammoniacale d’argent.
Pour transformer l’alloxantine en alloxane, on la réduit en poudre fine et on la chauffe au bain-marie avec 1 ½ partie d’eau. On ajoute goutte à goutte de l’acide nitrique concentré jusqu’à ce que toute l’alloxantine soit entrée en solution. On place la liqueur dans un exsiccateur d’acide sulfurique. Au bout d’un certain temps, l’alloxane se dépose sous forme de beaux cristaux incolores que l’on sèche sur du papier buvard ou sur une assiette poreuse.
1. Formation de muréxide. Une petite quantité d’alloxane est dissoute dans quelques gouttes d’eau et prudemment évaporée à sec sur une lame de platine. Il reste une tache rouge que l’ammoniaque fait virer au pourpre.
2. Formation d’acide alloxanique. Si, à une solution aqueuse d’alloxane, on ajoute un excès d’eau de baryte, on obtient un précipité blanc d’alloxanate de baryte.
56. Quinoléine C9H7N.
On mélange avec précaution dans un ballon de 1l,5 de capacité 24g de nitrobenzène, 38g d’aniline, 120g de glycérine et 100g d’acide sulfurique concentré. Après avoir muni le ballon d’un réfrigérant ascendant, on le chauffe doucement jusqu’à ce que le mélange entre en réaction. On éloigne alors la flamme. Lorsque l’ébullition s’est calmée, on recommence à chauffer, et l’on supprime la flamme dès que la réaction devient trop vive. Après quelques alternatives semblables, la réaction se poursuit sans trop de violence. On fait ensuite bouillir pendant 2 heures. On dilue avec de l’eau et l’on chasse le nitrobenzène par distillation avec les vapeurs d’eau. La solution est saturée par la soude caustique et soumise de nouveau à la distillation avec les vapeurs d’eau ; celles-ci entraînent la quinoléine et l’aniline. Pour se débarrasser de cette dernière, on ajoute de l’acide chlorhydrique en excès, puis du nitrite de sodium jusqu’à odeur persistante d’acide nitreux, même après agitation. On chauffe à l’ébullition jusqu’à destruction complète du diazobenzène formé, c’est-à-dire jusqu’à cessation du dégagement gazeux. La liqueur est de nouveau saturée par la soude caustique et soumise à la distillation avec les vapeurs d’eau. Le produit de distillation est secoué énergiquement avec de l’éther. Ce dernier, par distillation, abandonne la quinoléine qu’on sèche sur de la potasse caustique et qu’on distille. Son point d’ébullition est situé à 237°. Rendement : 40g environ.
Si l’on dissout quelques gouttes de la base dans de l’acide chlorhydrique et qu’on ajoute à cette solution un peu de chlorure de platine, on obtient un précipité jaune orange de chloroplatinate, qui, dissous dans de l’acide chlorhydrique dilué et chaud, cristallise en aiguilles rouges.
57. Éthers des acides dihydrocollidinedicarbonique et collidinedicarbonique.
On mélange, dans un verre à précipiter, 13g d’aldéhyde-ammoniaque fraîchement préparée avec 50g d’acétylacétate d’éthyle et l’on chauffe à feu nu. L’aldéhyde-ammoniaque se dissout et la liqueur se trouble bientôt par formation d’eau. On continue à chauffer jusqu’à faible ébullition, et l’on éloigne la flamme dès que la réaction devient trop vive. On recommence à chauffer lorsque l’ébullition se calme. La réaction peut être terminée en 5 minutes. La liqueur, troublée par un grand nombre de gouttes d’eau, s’épaissit. On ajoute au liquide huileux encore chaud, tout en le remuant, son volume égal d’acide chlorhydrique dilué. Au bout de peu de temps, l’huile se prend en une masse cristalline blanche. On filtre. Les cristaux sont lavés avec de l’acide chlorhydrique dilué, puis avec de l’eau, et pressés dans du papier buvard. Ils sont recristallisés dans la plus petite quantité possible d’alcool bouillant. Cette première cristallisation donne 27g de dihydrocollidine-dicarbonate d’éthyle presque pur. En concentrant les eaux mères, on récupère encore 7g d’un produit moins pur.
Pour transformer le dihydrodérivé en collidine-dicarbonate d’éthyle, on le réduit en poudre fine et on l’introduit dans son poids d’alcool. On dirige dans le mélange, soigneusement refroidi par de l’eau, un courant d’anhydride nitreux (obtenu par l’action de l’acide nitrique sur l’anhydride arsénieux) jusqu’à ce qu’on obtienne une solution claire et qu’une prise d’essai se dissolve complètement dans l’acide chlorhydrique dilué. L’alcool est évaporé. Il reste une huile qu’on secoue énergiquement avec un excès d’une solution diluée de carbonate de soude. On extrait par l’éther. Après distillation de l’éther, l’huile qui reste est séchée par du carbonate de potasse, puis distillée. Le collidine-dicarbonate d’éthyle passe vers 305°. Le rendement en produit distillé pur atteint les 80 pour 100 du dihydrodérivé employé.
Pour la transformation de l’éther en acide collidinedicarbonique, voyez Ann., t. CCXV, p. 26.
58. Méthylindol (Méthylcétol) C9H9N.
On mélange 30g de phénylhydrazine avec un peu plus que la quantité calculée d’acétone du commerce (point d’ébullition 56° à 58°) ; 18g d’acétone suffisent généralement. Le mélange s’échauffe fortement et il se sépare au bout de peu de temps une grande quantité d’eau. Pour achever la réaction, on chauffe 30 minutes au bain-marie et l’on traite une goutte de la solution par la liqueur de Fehling. Si celle-ci est fortement réduite, cela prouve qu’on se trouve en présence de phénylhydrazine non transformée. Il faudra donc rajouter une certaine quantité d’acétone, jusqu’à ce que la solution ne réduise presque plus la liqueur de Fehling.
La substance huileuse obtenue, rendue trouble par les gouttelettes d’eau, est versée dans un grand creuset en cuivre, qu’on chauffe 30 minutes environ au bain-marie, pour chasser l’excès d’acétone. On ajoute au résidu 200g de chlorure de zinc (du commerce) sec et l’on chauffe de nouveau au bain-marie, en ayant soin d’agiter continuellement la masse pour obtenir un mélange aussi intime que possible.
On met le creuset dans un bain d’huile qu’on chauffe jusqu’à 180. Après un séjour de quelques minutes à cette température, la substance prend une couleur foncée et l’on ôte le récipient du bain. Si l’on continue à agiter, la réaction se termine en peu de temps ; la coloration et le dégagement de vapeurs permettent de la suivre. Le produit est foncé. On le chauffe au bain-marie avec trois ou quatre fois son poids d’eau et un peu d’acide chlorhydrique chargé de dissoudre le chlorure de zinc. On soumet le tout à la distillation avec les vapeurs d’eau. Le méthylindol, entraîné lentement, mais complètement, sous forme d’une huile légèrement colorée en jaune, se solidifie bientôt. On filtre. Le produit brut est fondu encore une fois et, après solidification, séché avec du papier à filtrer, puis distillé. Rendement : 20g.
Le méthylindol doit être conservé dans des flacons fermant hermétiquement ou, de préférence, dans des tubes fermés au chalumeau.
On chauffe dans une éprouvette quelques fragments de méthylindol avec un peu d’eau, on présente aux vapeurs qui se dégagent un morceau de bois de pin, préalablement humecté d’acide chlorhydrique concentré. Il prend une coloration rouge intense.
59. Biphényle ou diphényle C6H5.C6H5.
On l’obtient en soumettant les vapeurs de benzène à la température du rouge. Dans un ballon S (fig. 17) de 1l,5 de capacité, placé sur le bain-marie, on verse 500g de benzène. On ferme le col du ballon par un bouchon percé de trois trous. Dans le premier, on met un tube de sûreté a. À travers le deuxième, passe le tube b, qui est relié au tube en fer forgé[12] R ; celui-ci, de 1m de longueur et de 20mm de diamètre, est rempli de petits morceaux de pierre ponce et placé dans un fourneau à combustion permettant de chauffer au rouge vif. Les vapeurs de benzène, sortant du ballon, arrivent dans le tube chauffé et y sont transformées en biphényle, en hydrogène et en d’autres produits. Le benzène non transformé et le biphényle qu’il dissout passent par le tube e et le réfrigérant K et retournent dans le ballon S à travers le tube c, dont l’extrémité plonge dans le benzène. Le tube d permet à l’hydrogène de s’échapper de l’appareil. On fait fonctionner l’appareil pendant 6 à 10 heures.

Le contenu du ballon se compose d’une solution assez concentrée de biphényle dans le benzène. On chasse ce dernier au bain-marie et l’on soumet le résidu à une distillation fractionnée. La fraction passant au-dessus de 150° se solidifie par refroidissement et consiste en biphényle presque pur, qu’on purifie par cristallisation dans l’alcool. Le rendement est en raison directe de la température du tube.
Si le gaz brûle sous une faible pression, le fourneau à combustion ordinaire ne suffit pas. Il sera alors préférable de se servir d’un fourneau de Fletcher ou d’un fourneau à charbon. Si toute l’opération a été bien conduite, le rendement peut dépasser 100g.
60. Benzoylacétone C6H5.CO.CH2.CO.CH3.
On dissout dans un ballon, à col court mais large, 4g de sodium dans 60g d’alcool absolu. On chauffe cette solution au bain d’huile pour chasser l’excès d’alcool, puis on laisse monter la température du bain jusqu’à 200°. En même temps on fait passer lentement un courant d’hydrogène absolument sec. On obtient ainsi sous forme d’un gâteau blanc et sec l’éthylate de sodium dont on a besoin pour la préparation. Il se laisse détacher des parois du ballon et concasser assez finement avec une baguette de verre pour qu’on puisse le retirer. Si l’on n’y parvient pas, il ne reste qu’à briser le ballon. L’éthylate de sodium est rapidement pulvérisé dans un mortier et remis aussi vite que possible dans le ballon, où l’on introduit peu à peu 35g d’éther acétique sec. Pendant cette opération, il faut avoir soin de refroidir le ballon avec de l’eau froide. Après 15 minutes environ, on ajoute 20g d’acétophénone. La cristallisation de la benzoylacétone sodée commence aussitôt. On verse de l’éther et on laisse reposer quelques heures pour que la combinaison sodée se dépose complètement. On filtre et on lave à l’éther, jusqu’à ce que celui-ci passe incolore. Le produit est séché à la température ordinaire et dissous dans l’eau froide. Par addition d’acide acétique, la benzoylacétone se dépose à peu près pure sous forme de cristaux presque incolores. Rendement : 19g environ.
1. Une solution de benzoylacétone dans l’alcool dilué donne avec le chlorure ferrique une coloration rouge intense ; avec l’acétate de cuivre, un précipité vert cristallin, qui est le sel de cuivre.
2. La benzoylacétone se dissout facilement à froid dans la soude caustique. Mais, si l’on chauffe, il y a décomposition et formation d’acétophénone, qui se sépare en gouttelettes huileuses, si la solution n’est pas trop diluée.
61. Benzophénone C6H5.CO.C6H5.
On mélange 100g d’acide benzoïque avec un peu plus que la quantité calculée de chaux éteinte. On ajoute 1l d’eau et l’on chauffe à l’ébullition, jusqu’à ce que tout l’acide soit dissous ; il faut que la solution reste alcaline. Pour se débarrasser de l’excès de chaux, on filtre à l’ébullition. Par refroidissement, le benzoate de calcium se dépose en grande partie sous forme d’aiguilles blanches. Le reste peut être récupéré par concentration des eaux mères. Le sel est filtré à la trompe, bien pressé dans du papier à filtrer et complètement desséché dans des capsules de métal chauffées à feu nu.
On introduit ce sel bien sec dans une cornue en fer ou en cuivre reliée par un large tube de verre à un réfrigérant descendant. Le produit ne doit pas occuper plus des deux tiers de la cornue. On chauffe au moyen d’une forte flamme de façon à obtenir aussi vite que possible une distillation sèche du sel. Au commencement, il passe un mélange brun clair composé de benzène, de benzophénone et de produits empyreumatiques. On arrête l’opération lorsque le produit de distillation devient brun foncé et visqueux. Le mélange obtenu est séché par du chlorure de calcium et soumis à la distillation fractionnée. La partie passant entre 250 et 310° contient la benzophénone ; elle se solidifie en général au bout de peu de temps ; mais il arrive aussi parfois qu’elle conserve pendant plusieurs jours une consistance sirupeuse. Dans ce cas, on amorcera la cristallisation en jetant un petit cristal de benzophénone. Pour débarrasser les cristaux de leur eau mère huileuse, on les presse entre deux feuilles de papier buvard, ou on les étend sur une assiette poreuse. On les recristallise dans la ligroïne. Rendement : 15g à 20g.
La benzophénone bout à 305°.
62. Benzophénone-oxime

10g de benzophénone pure et 12g de chlorhydrate d’hydroxylamine sont dissous dans un mélange de 130cm³ d’alcool et de 80cm³ d’eau. On y ajoute une solution de 2g de soude caustique dans 30cm³ d’eau et l’on chauffe le tout pendant 1 heure au réfrigérant ascendant.
Le produit de réaction est étendu de beaucoup d’eau et acidulé par de l’acide sulfurique dilué. La benzophénone-oxime se dépose en fins cristaux.
Après plusieurs heures, l’oxime est filtrée à la trompe, lavée à l’eau et séchée. Le rendement est presque quantitatif et la benzophénone-oxime est généralement assez pure pour n’avoir pas besoin d’être recristallisée. Point de fusion : 141°.
3g de benzophénone-oxime sont dissous dans 40cm³ d’éther complètement desséché sur du sodium. À cette solution on ajoute, par petites portions, 4g de perchlorure de phosphore très finement pulvérisé.
L’éther est distillé. Le résidu huileux, qui se compose d’oxychlorure de phosphore et de chlorhydrate de benzanilide, est trituré dans une capsule avec de la glace pilée. L’huile se solidifie en un produit granuleux qu’on broie ; il est filtré à la trompe, lavé à l’eau et séché. La transformation est quantitative. La benzanilide formée (C6H5.CO.NH.C6H5), recristallisée de l’alcool bouillant, est alors tout à fait pure. Elle fond à 162°.
63. Phénanthrène-quinone C14H8O2.
On dissout 30g de phénanthrène aussi pur que possible dans 120g d’acide acétique bouillant. On y ajoute peu à peu une solution de 70g d’acide chromique dans 200g d’acide acétique. (On dissout d’abord l’acide chromique dans très peu d’eau et l’on verse cette solution dans l’acide acétique.) Le mélange s’échauffe de lui-même et l’on s’arrange à ce que sa température se maintienne aux environs de 100°-110° (point d’ébullition de l’acide acétique glacial). Pour finir, on distille la majeure partie de l’acide acétique et l’on ajoute de l’eau au résidu. Il se dépose une masse cristalline orangée, qui contient la phénanthrène-quinone et un peu de phénanthrène non transformé. On filtre et on lave avec un peu d’eau chaude. Pour purifier le produit brut, on le traite à chaud par une solution diluée de bisulfite de soude. On filtre et l’on acidule la liqueur par de l’acide sulfurique, ce qui précipite la quinone. On la cristallise enfin dans beaucoup d’alcool bouillant.
64. Triphénylméthane CH(C6H5)3.
On mélange 500g de benzène pur et sec avec 100g de chloroforme. On chauffe modérément et l’on ajoute par petites portions 150g de chlorure d’aluminium sublimé et pulvérisé[13]. Le ballon est muni d’un réfrigérant ascendant et chauffé pendant 2 heures à l’ébullition. On verse le liquide, avec précaution et en agitant continuellement, dans son volume d’eau glacée, puis on ajoute de l’acide chlorhydrique jusqu’à dissolution complète des composés d’aluminium. Au moyen d’un entonnoir à robinet, on ne garde que la couche benzénique, qu’on verse sur un filtre à plis bien sec pour la débarrasser de l’eau qu’elle garde en suspension. Le benzène est distillé au bain-marie. Toutes ces opérations, vu la grande inflammabilité des vapeurs de benzène, doivent être conduites avec une extrême prudence. Le résidu est introduit dans un ballon à distiller garni de papier d’amiante et soumis à la distillation fractionnée. On recueille ce qui passe au-dessus de 150°.
À 200°, il se produit un vif dégagement d’acide chlorhydrique provenant de la décomposition d’un chlorure complexe. Au-dessus de cette température, il distille un mélange de diphénylméthane et de triphénylméthane. On ôte le thermomètre et l’on continue la distillation, jusqu’à ce que le résidu commence à charbonner. Le produit de distillation brut est fractionné encore une fois. Le triphénylméthane distille au-dessus de 300° et se solidifie dans le flacon récepteur. Pour le purifier, on le dissout dans le benzène bouillant. Par refroidissement, il y a dépôt de beaux cristaux, qui sont une combinaison de benzène et de triphénylméthane, et qui sont recristallisés encore une fois du benzène, si c’est nécessaire. Si l’on chauffe ces cristaux au bain-marie, ils perdent facilement leur benzène. Le triphénylméthane fondu qui reste, dissous dans l’alcool bouillant, cristallise par refroidissement en prismes ou en paillettes incolores fondant à 93°. Le rendement dépend de la qualité du chlorure d’aluminium employé ; il varie entre 10g et 30g.
Une petite quantité de triphénylméthane (environ 0g,5) est dissoute à froid dans quelques centimètres cubes d’acide nitrique fumant. Par addition d’eau, le produit nitré précipite sous forme de flocons jaunes. Ceux-ci sont filtrés, dissous dans l’acide acétique bouillant et réduits par de la poudre de zinc. On dilue avec de l’eau et l’on filtre. La liqueur claire est saturée par de l’ammoniaque ; la base précipite ; on la recueille sur un filtre. Ce produit contient une grande quantité de paraleucaniline. Si l’on chauffe sur une lame de platine une prise d’essai avec de l’acide chlorhydrique concentré, on aperçoit, après évaporation de l’acide, la belle coloration de la fuchsine.
65. Triphénylcarbinol (C6H5)3C.OH.
On prend 1g,2 de fil de magnésium poli, qu’on coupe en morceaux de 1cm ou 2cm de longueur. On introduit ceux-ci dans un ballon à fond rond bien desséché, d’une capacité de 300cm³, et dans lequel on verse une solution de 8g de bromobenzène dans 40g d’éther desséché sur du sodium ; on jette dans le mélange une paillette d’iode. Le ballon, muni d’un réfrigérant et traversé par un courant d’hydrogène sec, est chauffé (prendre garde de ne pas approcher de flamme de l’ouverture du réfrigérant). De légers flocons paraissent, provenant de l’action sur le magnésium de traces d’humidité qu’on ne peut chasser complètement. Ces flocons disparaissent peu à peu et le magnésium entre en solution. Il faut environ 2 heures pour que tout le magnésium se dissolve ; lorsqu’il ne reste plus que quelques impuretés au fond du ballon, on laisse refroidir. On ajoute une solution de 9g,1 de benzophénone dans 25g d’éther absolument sec. Le liquide prend aussitôt une coloration, rouge clair, puis une liqueur épaisse et visqueuse se sépare. On réchauffe le ballon pendant une demi-heure environ, ce qui provoque une vive réaction et solidifie cette liqueur. On refroidit le ballon ; on y introduit de petits morceaux déglace et de l’acide sulfurique dilué. Il y a décomposition. Le mélange est soumis à la distillation avec les vapeurs d’eau jusqu’à ce que le liquide distillé passe clair. Les vapeurs d’eau entraînent l’éther et tous les produits accessoires formés (benzène, biphényle). Le triphénylcarbinol reste dans le ballon à l’état presque pur. Recristallisé du benzène, il fond à 159°. Rendement : 10g.
Le triphénylcarbinol se dissout dans l’acide sulfurique concentré en donnant une coloration d’un jaune intense. Si on le dissout dans l’acide acétique, on obtient une solution incolore, mais qui devient aussitôt d’un beau jaune dès qu’on y ajoute quelques gouttes d’acide chlorhydrique concentré.
66. Vert à l’aldéhyde. Leucobase C6H5.CH[C6H4N(CH3)2]2.
Dans une capsule en porcelaine, on mélange 20g d’aldéhyde benzoïque avec 50g de diméthylaniline et 40g de chlorure de zinc solide. On fait chauffer le tout pendant quelques heures au bain-marie, en remuant constamment. Si, pendant cette opération, la masse devenait trop dure, on la diluerait avec un peu d’eau. La réaction est terminée, lorsque l’odeur d’amandes amères a presque totalement disparu. Le produit est mis dans un ballon et additionné d’eau. On fait passer un fort courant de vapeurs d’eau qui entraîne la diméthylaniline non transformée. La leucobase, qui est verdâtre, est facilement séparée par décantation de la solution contenant le chlorure de zinc ; elle se prend, par refroidissement, en une masse dure qu’on dissout dans l’alcool absolu et bouillant. D’une solution diluée, la base se dépose par refroidissement en fines petites aiguilles presque incolores. Si la solution est concentrée, la base se dépose généralement en une masse amorphe, qui cristallise au bout d’un certain temps. Par cristallisations répétées, ou peut obtenir la leucobase tout à fait incolore. Le rendement peut atteindre les 90 pour 100 de la théorie.
On dissout 1 partie de leucobase dans 100 parties d’acide chlorhydrique, étendu de telle façon qu’on ait 1 molécule de la base pour 4 molécules d’acide chlorhydrique ; on refroidit la solution. On y ajoute, dans l’espace de 5 minutes et tout en agitant constamment, la quantité calculée de peroxyde de plomb finement pulvérisé et préalablement mélangé avec 6 parties d’eau. Après addition, on secoue encore 5 minutes, puis on filtre.
On ajoute à la solution de la matière colorante ainsi obtenue 2 molécules de chlorure de zinc, puis une solution concentrée et chaude de sel marin, jusqu’à ce qu’une prise d’essai ne colore presque plus un morceau de papier à filtrer.
Après refroidissement complet, on filtre la matière colorante qui s’est déposée. On la redissout dans le moins d’eau bouillante possible et on la précipite de nouveau par une solution de sel marin.
67. Fluorescéine et Éosine
C20H12O5 et C20H8O5Br4.
On chauffe dans un bain d’huile à 195°-200° un mélange de 10g d’anhydride phtalique pulvérisé et de 14g de résorcine du commerce, jusqu’à ce que le dégagement de vapeurs d’eau ait cessé et que la masse, qui était primitivement fluide, soit devenue tout à fait solide. Le produit est pulvérisé, bouilli avec de l’eau, puis filtré. On dissout la partie insoluble dans de la soude caustique étendue ; on recouvre cette solution d’une couche d’éther, et l’on précipite la fluorescéine par de l’acide sulfurique dilué.
Dans cet état, la fluorescéine est facilement soluble dans l’éther ; on la dissout dans celui-ci. Si, à la solution éthérée, on ajoute de l’alcool et qu’on distille l’éther, la fluorescéine se dépose en croûtes cristallines rouges, qui sont alors presque insolubles dans l’éther. La fluorescéine donne en solution ammoniacale une fluorescence verte remarquable. La formation de fluorescéine est employée comme réactif dans la recherche de l’acide phtalique ou de la résorcine.
Le rendement est presque quantitatif.
Pour transformer la fluorescéine en éosine, on mélange la fluorescéine finement pulvérisée avec 4 fois la quantité d’acide acétique. On y ajoute la quantité calculée de brome (4 molécules) dilué également dans 4 fois sa quantité d’acide acétique. Lorsqu’on chauffe, la fluorescéine se dissout. Par addition d’eau, l’éosine précipite en flocons rouges, qu’on sèche et qu’on cristallise dans beaucoup d’alcool bouillant.
68. Anthraquinone C14H8O2.
5g d’anthracène du commerce finement pulvérisé sont dissous dans 220cm³ d’acide acétique bouillant. La solution, filtrée si c’est nécessaire, est maintenue à l’ébullition, pendant qu’on lui ajoute une solution de 50g d’acide chromique dans 50cm³ d’acide acétique à 50 pour 100. On continue à chauffer jusqu’à ce qu’une prise d’essai, déposée sur une pièce d’argent, donne au bout de peu de temps une tache rouge (?). Après refroidissement on ajoute 750cm³ d’eau. Après plusieurs heures, on filtre le précipité formé, qu’on lave avec de l’eau, puis avec une solution de potasse caustique et de nouveau avec de l’eau. L’anthraquinone ainsi obtenue est sous forme de fines aiguilles jaunes. Pour des déterminations quantitatives, on n’emploie que 1g d’anthracène brut et l’on fait une correction au chiffre obtenu, pour tenir compte de l’anthraquinone restée en solution.
69. Alizarine C14H6O2(OH)2.
50g de soude caustique sont dissous à chaud dans 50g d’eau. On y ajoute 10g d’anthraquinone-monosulfonate de sodium et une solution concentrée de 3g de chlorate de potassium. La masse doit avoir à 120-130° une consistance pâteuse. Si ce n’est pas le cas, on évapore jusqu’à ce qu’on obtienne la consistance désirée. On prend alors un tube en fer forgé, muni d’un couvercle à pas de vis, et qui a été essayé à une pression de 20atm. Ce tube est rempli jusqu’aux deux tiers de sa longueur avec le produit de réaction et fermé hermétiquement. On le chauffe au bain d’huile pendant 20 heures à 175°-185°.
À la place du bain d’huile, on peut se servir d’un bain de vapeurs d’aniline d’une forme particulière et qui demande moins de surveillance.

a (fig. 18) est un récipient de cuivre de forme cylindrique, de 63cm de hauteur et 13cm de largeur et terminé par un fort rebord, consolidé au moyen d’un anneau de fer. Sur ce rebord, on peut fixer un couvercle b au moyen des vis c, en ayant soin d’intercaler un anneau de papier d’amiante pour rendre la fermeture plus hermétique. Le couvercle b est percé de 2 trous, laissant passer un thermomètre et un tube de verre, dans lequel se condenseront les vapeurs d’aniline. La partie inférieure du cylindre est munie d’une plaque de cuivre percée de trous et sur laquelle repose le tube d. On introduit dans le cylindre 200g à 250g d’aniline. Le couvercle est vissé et l’appareil est chauffé à feu nu. La température doit être maintenue pendant 20 heures à la température d’ébullition de l’aniline (184°).
On laisse refroidir jusqu’à 70° et l’on enlève le produit, d’abord mécaniquement, puis par de l’eau bouillante. L’alizarate de sodium est difficilement soluble dans l’eau chaude, aussi doit-on faire bouillir énergiquement et assez longtemps.
On sature la solution par de l’acide sulfurique dilué, on la soumet avec le précipité à l’ébullition pendant un quart d’heure et, lorsqu’elle s’est refroidie à 70°, on la filtre. Le précipité est lavé à l’eau chaude, jusqu’à ce que les eaux de lavage ne contiennent plus d’acide sulfurique.
Si l’on veut obtenir l’alizarine cristallisée en aiguilles ; on la sublime entre deux verres de montre au bain de sable à 300°.
70. Camphre-oxime C10H16NOH.
10g de camphre sont dissous dans 150g d’alcool. On ajoute à cette liqueur une solution aqueuse concentrée de 10g de chlorhydrate d’hydroxylamine, puis une solution également concentrée contenant 15g de soude caustique. Le tout est chauffé au bain-marie. Il est quelquefois nécessaire d’ajouter une nouvelle quantité d’alcool pour dissoudre tout le camphre. La réaction est terminée lorsqu’une prise d’essai, diluée avec de l’eau, ne donne plus de précipité, ce qui a lieu généralement au bout d’une heure. La solution alcoolique est alors étendue de beaucoup d’eau, filtrée s’il y a lieu, pour se débarrasser d’un léger précipité floconneux, et additionnée d’acide acétique, jusqu’à faible réaction acide. La camphre-oxime se dépose en une masse cristalline incolore. Le rendement atteint 75 pour 100 de la théorie. Pour purifier le produit, on le recristallise dans l’alcool dilué.
- ↑ Le point de fusion déterminé d’après cette méthode sera trouvé un peu trop bas parce qu’une petite partie seulement de la colonne mercurielle du thermomètre plonge dans le bain. Cependant la plupart des points de fusion sont obtenus de cette manière et reproduits dans la littérature avec la mention « non corrigés ». Pour avoir le vrai point de fusion, il faut dresser pour chaque thermomètre une table de correction indiquant pour un nombre de degrés assez grand les différences qu’il présente avec les thermomètres courts de Zincke. Cette opération se fera d’après la disposition de la figure 6.
- ↑ Il faut établir préalablement la teneur du nitrite de soude commercial par titration avec du permanganate de potasse ; on peut aussi, ayant quelque pratique, opérer avec une quantité indéterminée de NaNO2, en reconnaissant la fin de la diazotation par l’essai à la touche sur le papier iodoamidonné. Tant que la solution contient de l’aniline non transformée, une goutte, additionnée d’une solution d’acétate de soude, donne, après peu de temps, un trouble provenant de la formation de diazoamidobenzène.
- ↑ On le prépare en additionnant environ 20g de l’acide fumant commercial, contenant 70 pour 100 SO3, à 130g d’acide concentré.
- ↑ Il faut, autant que possible, calculer auparavant les quantités des réactifs. Cela se fait le plus commodément lorsque la concentration des solutions employées d’acides, d’alcalis et de sels est établie d’après les proportions normales simples.
- ↑ Tous les liquides dont le point d’ébullition est situé en dessous de 100° peuvent être distillés sur le chlorure de calcium.
- ↑ Provenant d’une bombe ou obtenu en chauffant une solution concentrée d’ammoniaque et séché par le passage sur de la chaux vive.
- ↑ La plupart des fusions potassiques peuvent s’effectuer avec des creusets et spatules en cuivre dont le prix beaucoup plus bas les fera préférer aux ustensiles d’argent.
Pour déterminer la température de la fusion, on se sert de thermomètres ordinaires, protégés contre l’action de l’alcali par une gaine étroitement ajustée en tôle de cuivre mince. Lorsqu’ils sont suffisamment forts, ils peuvent être utilisés comme agitateurs.
- ↑ Pour préparer l’amalgame, on verse 500g de mercure sec et pur dans un mortier de porcelaine et l’on ajoute assez rapidement les 2,5 pour 100 de sodium métallique découpé en disques de la grosseur d’une pièce de 2 francs. Il faut avoir soin de maintenir avec le pistil chaque morceau au fond du mortier. De cette manière et en opérant avec quelque précaution, on évite l’inflammation si désagréable du métal, ainsi que la projection de particules enflammées de sodium. Il est toutefois nécessaire pendant cette opération de se placer derrière la vitre de la hotte et de se protéger les mains par des gants.
On conserve l’amalgame dans des flacons à large ouverture bouchés à l’émeri et munis de protège-bouchons.
- ↑ Le sulfate de méthyle étant inodore et vénéneux, il faut se garder d’en respirer les vapeurs. On travaillera donc sous la hotte jusqu’à complète destruction du sulfate de méthyle.
- ↑ Si l’on n’a pas à sa disposition une solution aqueuse d’anhydride sulfureux, on se sert, pour la préparation de ce gaz, d’une solution de bisulfite de soude du commerce, dans laquelle on laisse tomber goutte à goutte, d’un entonnoir à robinet (à brome), de l’acide sulfurique concentré. On peut facilement régler le courant d’anhydride sulfureux qui se dégage.
- ↑ On obtient l’acide urique en chauffant à l’ébullition dans un ballon à fond rond, jusqu’à presque complète dissolution, 25g d’excréments de serpents avec une solution très diluée de soude caustique (50g à 40 pour 100 dans 500g d’eau). On filtre à chaud et l’on acidule par de l’acide chlorhydrique.
- ↑ Comme on les emploie pour les conduits de gaz.
- ↑ Il ne faut ouvrir qu’avec grande précaution le flacon renfermant le chlorure d’aluminium, à cause de la pression qui y existe souvent.